


Introduction
The temporal distribution of environmental tracer substances can be determined by analyzing successive snow layers of ice cores sampled in the ice caps. Such a study constitutes an effective method to:
identify the principal and secondary sources of atmospheric aerosols such as sea spray, marine biogenic, volcanic, crustal or anthropogenic inputs;
understand the transport processes of environmentally relevant substances, with particular attention to sea/ atmosphere/snow interface exchange;
highlight particular events capable of putting into the atmosphere high quantities of well-defined compounds (e.g. volcanic eruptions, biomass-burning, nuclear experiments or accidents).
In particular, volcanic input is one of the main aerosol sources and the most important localised geochemical source. Volcanic eruptions can introduce into the atmosphere significant quantities of H2SO4, HCl, HF and HBr (10, 8, 0.4 and 0.08 × 1012 g a-1, respectively; Reference BrimblecombeBrimblecombe, 1996), and, in the case of an explosive eruption, gas and particulate can be injected into the highest part of the troposphere or directly into the stratosphere, with high half-life times and global-scale diffusion (Reference Hammer, Clausen and DansgaardHammer and others, 1980; Reference Legrand and DelmasLegrand and Delmas, 1987; Reference Clausen and HammerClausen and Hammer, 1988; Reference Delmas, Kirchner, Palais and PetitDelmas and others, 1992; Reference Aristarain and DelmasAristarain and Delmas, 1998).
In areas characterised by low crustal and anthropogenic contributions, such as Antarctica, volcanic emissions can constitute a fundamental input to the atmospheric aerosol composition. The gas-phase substances and particulate content can play a relevant role in environmental and climate variations, by means of sunlight-scattering processes and formation and growth of cloud-condensation nuclei (Reference Hammer, Clausen and DansgaardHammer and others, 1980; Reference HandlerHandler, 1989; Reference Rampino and SelfRampino and Self, 1992; Reference Zreda-Gostynska, Kyle, Finnegan and PrestboZreda-Gostynska and others, 1997). Besides, the identification of historically known volcanic events in ice cores is the main technique for obtaining reference horizons for a reliable absolute dating of the sampled snow layers (Reference Langway, Clausen and HammerLangway and others, 1988; Hammer, 1989; Reference Delmas, Kirchner, Palais and PetitDelmas and others, 1992). Volcanic levels can be identified mainly by high and sudden increases of specific markers such as sulphate, fluoride, bromide, chloride (from the related acids), carboxylic acids, ash and total acidity, and of non-specific parameters, such as dielectric properties and electrical conductivity (Reference Hammer, Clausen and DansgaardHammer and others 1980; Reference Palais, Kirchner and DelmasPalais and others, 1990; Reference Aristarain and DelmasAristarain and Delmas, 1998).
In this paper we report the chemical characterisation by ion chromatographic (IC) analysis of a volcanic event recorded in an ice core drilled on Styx Glacier plateau (1750 m a.s.l.), northern Victoria Land, East Antarctica. The volcanic event was shown by a dark layer at 93.41 m depth and dated to about AD 1500.
Experimental
Sampling station
The ice-core sampling station, Styx Glacier plateau (163°56’E, 73°52’S; 1750 ma.s.L), is shown in Figure 1. The Styx Glacier plateau is a 150 km2 plateau, located between the Campbell Glacier and Tinker Glacier valleys, about 50 km from the coastline (Wood Bay) and at 1600-1800 m a.s.1. This area was chosen by satellite observations of snow morphology in the area of Terra Nova Bay (about 100 000 km2 in northern Victoria Land), which were used to isolate stations where wind-redistribution effects (shown by blue ice, sastrugi or snowdrift areas) are as low as possible (Stenni and others, unpublished). In fact, several authors (e.g. Reference Pettre, Pinglot, Pourchet and ReynaudPettre and others, 1986; Reference GoodwinGoodwin, 1990) have pointed out the importance of katabatic winds for the net annual snow-accumulation rate. Transport (ablation, accumulation and redistribution) processes can change the snow-layer sequence, making the reconstruction of successive depositions impossible. The Styx Glacier plateau wind-field analysis indicated that the Campbell Glacier valley channels the katabatic winds coming from the west and northwest, so that only moderate snowdrifts (west-east oriented) are present.
Fig. 1. Map of sampling station: Styx Glacier plateau, northern Victoria Land, Antarctica (163° 56′ E, 73° 52′ S; 1750 m a.s.l.).
At this station, snow and firn samples were collected in snow pits and shallow firn cores during several Italian Antarctic Campaigns. The samples were analyzed to determine the chemical composition of snow and the mean snow-accumulation rate. These results are reported in Reference Piccardi, Udisiti and CasellaPiccardi and others (1994a, b, 1996a, b) Reference UdistiUdisti (1996) and Reference Udisti, Traversi, Becagli and PiccardiUdisti and others (1998b) with the following conclusions:
-
(1) a well-defined seasonal signal for H2O2, non-sea-salt sulphate (nssSO4 2-), methanesulphonate (MSA) and δ180 is present, and therefore such compounds can be used as seasonal markers (high summer values). This evidence confirms an undisturbed snow-layer sequence;
-
(2) ice crusts showing summer snow melting are missing, due to the rather high elevation (about 1700 m a.s.1.);
-
(3) a fairly high mean annual accumulation rate of about 160 kg m-2 a-1 w.e. allows a good temporal resolution of the sampled snow layers.
For the above reasons, the Styx station was chosen as a site for medium-depth ice-coring in the 1995-96 Italian Campaign.
Ice-core sampling
The Styx Glacier ice core (116 m deep) was drilled in November 1995 using a wire-driven corer ICE-DRILL 4 (FS INVENTOR AG, Switzerland). This drill collects 107 mm diameter ice cores with a maximum length of 950 mm per run, operating at a maximum depth of 150 m. The single ice-core sections obtained step by step were externally cleaned on site and closed in double-sealed polyethylene bags. The ice-core sections were weighed to determine their density; the values range from 0.38 gcm-3 (superficial layers) to 0.73-0.98 g cm-3 for layers deeper than 80 m. During the ice-core collection and handling, care was taken to minimise contamination (clean-room clothes, facial masks and polyethylene gloves).
Visual examination of an ice-core section showed a thin, dark layer (about 10 mm thick) located at 93.41 m. An interval of 300 mm around the dark layer was immediately subsampled and analyzed. This ice-core section was divided into ten subsamples with a resolution of 30 mm.
All decontamination (by mechanical removal of a 10 mm external layer with a hand scraper) and cutting operations were performed in a cold room under a class 100 laminar hood. Each sample manipulation was previously controlled by a similar operation performed on a synthetic ice core obtained from ultra-pure water to minimise the contamination. The operational blank shows contamination levels 1-2 orders of magnitude lower than the lowest measured concentrations ("background" level measured on the samples far away from the dark layer).
Ice-core approximate dating
At the time of writing, the subsampling of the remaining ice core is still in progress, so an accurate dating of the ice core is not yet available. An approximate dating was estimated using the firn-densification model of Reference Herron and LangwayHerron and Langway (1980) to model the density data obtained from the weight and area measurements on all the ice-core sections (Reference LingLing, 1985). Figure 2a shows the experimental density/depth profile. These data have been fitted on the basis of the following equation proposed by Reference Herron and LangwayHerron and Langway (1980):
where ρ and x are the density and depth, respectively. ρo, ρm and L are the calculated parameters of the fitting: ρo (0.41 g cm"3) is the density of surface snow, ρm (0.87 gcm-3) is the maximum density in the dry-snow zone and L (27) is a parameter characteristic of the sampling site. Considering a mean annual accumulation of 160 kg m-2 a-1 w.e. and taking into account the relationship between density, accumulation rate and depth, it is possible to plot the age/depth curve as shown in Figure 2b. This curve has been used to extrapolate the age of the ice-core section in which the volcanic layer was found.
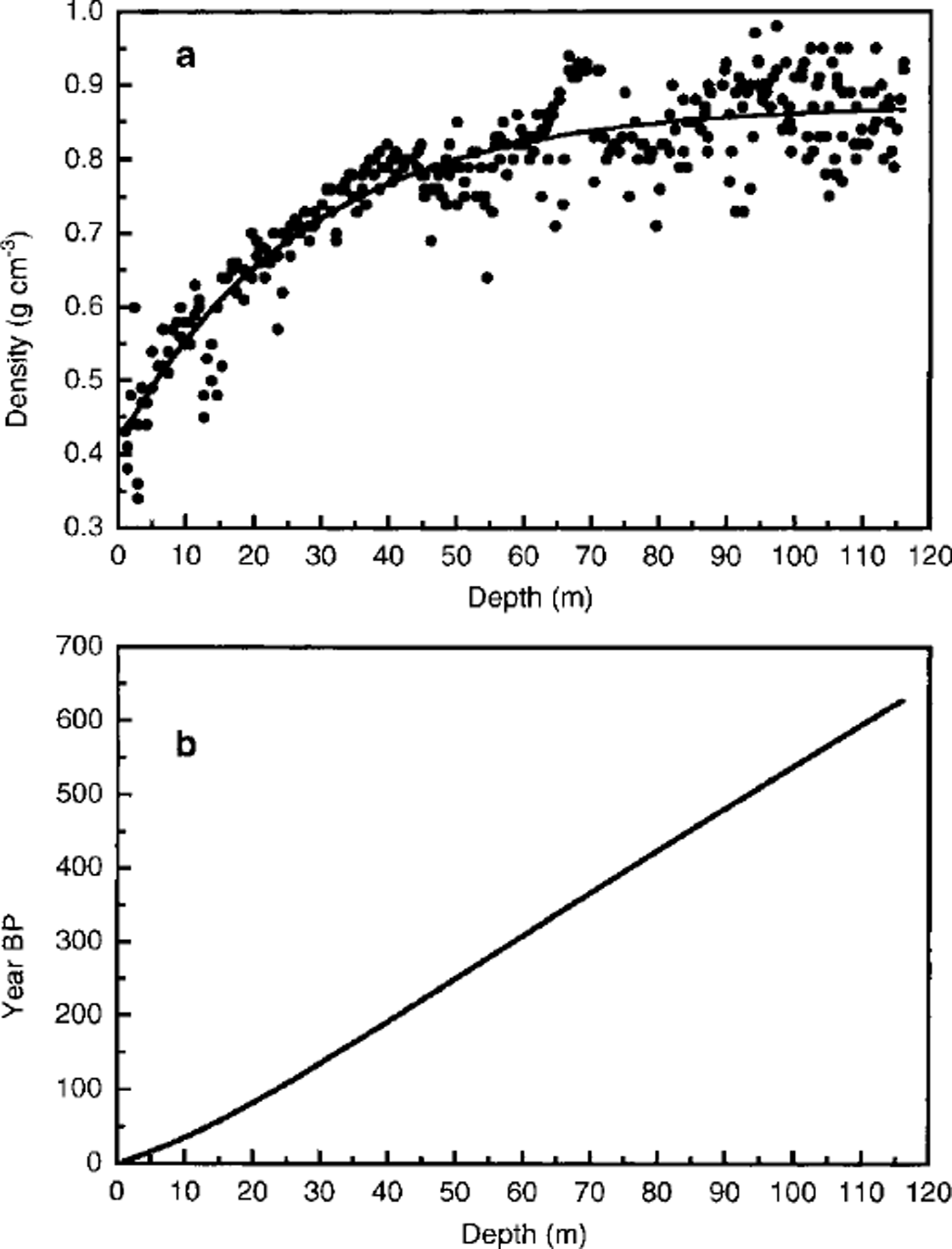
Fig. 2 Ice-core dating by the firn-densification model of Reference Herron and LangwayHerron and Langway (1980) to model the density data (Reference LingLing, 1985). (a) Experimental density/depth profile, (b) Age/depth curve obtained considering a mean annual accumulation of 160 kg m–2 a–1 w. e. This curve has been used to extrapolate the age of the ice-core section in which the volcanic layer was found (93.41m depth).
According to this model, the whole ice core covers approximately the period AD 1995-1350, so the oldest layers (depth 116 m) can be attributed to snow precipitation about 650 years ago. The age of the dark layer, located at 93.41 m depth, can be estimated at about 500 ± 20 years (around AD 1500).
Chemical analysis
The subsamples were melted in their closed containers, and IC measurements were performed immediately after melting to avoid gas-phase contaminant uptake (above all, am monia and carboxylic acids; Reference Udisti, Barbolani and PiccardiUdisti and others, 1991, Reference Udisti, Bellandi and Piccardi1994). At the IC injection time, the samples were filtered on a 0.45 μm teflon membrane, chosen as a conventional limit between the soluble and insoluble fractions. Therefore, the chemical analysis is related only to the soluble components (particle dimension <0.45μm).
IC methods for inorganic anions, cations and some organic anions (plus fluoride) are reported elsewhere (Reference Udisti, Barbolani and PiccardiUdisti and others, 1991,Reference Udisti, Bellandi and Piccardi1994,Reference Udisti, Becagli, Traversi, Vermigli and Piccardi1998a, Reference Udisti, Traversi, Becagli and Piccardib). Table 1 summarises the techniques used for the analyzed compounds and the detection limits.
Table 1. IC parameters for determination of inorganic anions, cations and some organic anions
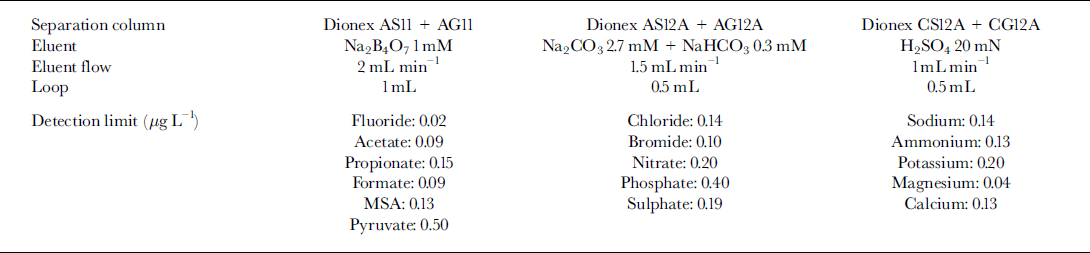
Figure 3 shows examples of the IC separations and identification of inorganic anions (Fig. 3a-b), and organic (plus fluoride) anions (Fig.3c-d) for the more concentrated subsamples (dark layer 93.41 m depth).

Fig. 3. Ion chromatograms for sample 7 (93.41m depth) of inorganic anions without dilution (a) and at 1:40 dilution (b); and organic anions plus fluoride without dilution (c) and at 1:40 dilution (d). 1. Chloride, 2. Nitrite, 3. Bromide, 4. Nitrate, 5. Phosphate, 6. Sulphate, 7. Fluoride, 8. Acetate, 9. Propionate, 10. Formate, 11. MSA, 12. Pyruvate. Concentration values are given inTable 2.
Data Discussion
The concentration values for the determined substances are reported in Table 2. A series of numbers (the youngest, the dark-layer and the oldest subsamples are labelled 1, 7 and 10, respectively) identifies the ten subsamples. Also inTable 2 the depth of each subsample is reported. Figure 4 shows the concentration profiles of total sulphate (totS042-), nssSO 2- and fluoride. The nssSO4 2- was calculated from the sodium content, used as a sea-spray marker, by
where swSO42- means sea-water sulphate and 0.253 is the S04 2- /Na+ ratio (w/w) in sea water.
Table 2. Concentration data for the ten subsamples around the volcanic layer (No. 7) for the Styx Glacier ice core
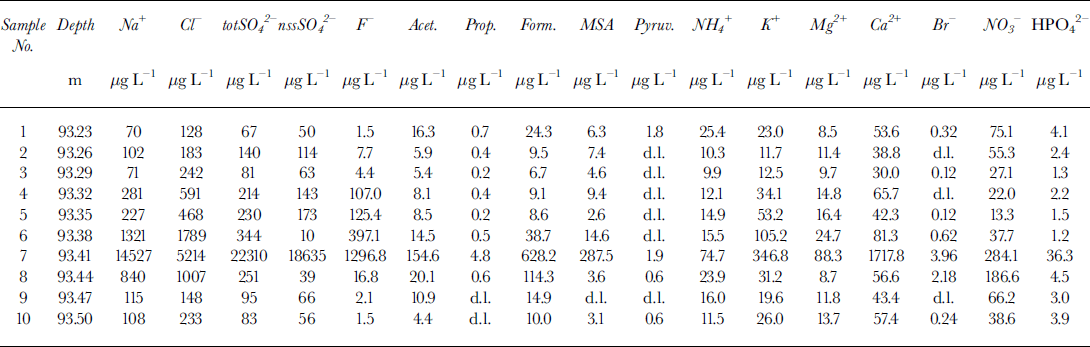

Fig. 4. Concentration/depth profiles of total sulphate (a), non-sea-salt sulphate (b) and fluoride ( c) in the ten subsamples around the volcanic layer from 93.23 (sample 1) to 93.50 (sample 10) m depth.
Very high concentration peaks of sulphate and fluoride for the dark-layer sample (No. 7) show how these substances can be useful for the identification of volcanic events thanks to the massive emission of HF and SO2 (successively oxidised to H2SO4). Fluoride and sulphate peaks, together with high values of acidity and conductivity, were detected in snow and ice layers recording volcanic eruptions from Greenland and Antarctica (Delmas and others, Reference Delmas, Legrand, Aristarain and Zanolini1985, Reference Delmas, Kirchner, Palais and Petit1992; De Angelis and Legrand, Reference De Angelis and Legrand1994; Zielinski and others, Reference Zielinski, Dibb, Cv, Mayewski, Whitlow and Twickler1997; Zreda-Gostynska and others, Reference Zreda-Gostynska, Kyle, Finnegan and Prestbo1997; Reference Aristarain and DelmasAnstarain and Delmas, 1998). The concentrations of totSO42- and nssSO42- (Fig. 4a and b) are very similar, so practically all the sulphate content can be ascribed to H2SO4. In sample 7, the concentration values for nssSO42- and F- are 370 and 860 times higher, respectively, than in the ice-core-section extremities (samples 1 and 10). The nssSO42- concentration in sample 7 is about 220 times higher than the mean value (86.5 Mg L-1) measured in the same station by a 2.5 m snow pit (covering the period 1988-93) dug during the 1993-94 Italian Antarctic Campaign (Table 3). Referring to the same dataset, the fluoride levels in the dark layer are about 1200 times higher than the mean 1988-93 value (0.99 Mg L-1). Detailed examination of the concentration profiles of fluoride (Fig. 4c), the marker showing the largest increase and the least influence by other source contributions, shows a typical atmospheric scavenging trend: the volcanic event starts with sample 8, where a concentration one order of magnitude higher than the mean was measured (16.8 μg L-1), and reaches a maximum in sample 7. In successive samples, the F- concentration quickly decreases, with a hyperbolic trend up to sample 1 in which it reaches the same concentration as sample 10.
Table 3. Statistical concentration values for snow-Pit samples (1988-93) collected in the Italian Antarctic Campaign 1993-94
The nssSO42- behaviour is similar, with the same low concentration in samples 1 and 10, and a maximum in sample 7, but the decrease is not constant, showing values 2-3 times higher than the background (samples 1 and 10), alternating with low values (samples 6 and 3). Further contributions, naturally variable, of sulphate and above all, of Na+ (sample 6) used for the ussSO42- calculation can mask the observation of the scavenging processes of volcanic nssSO4 2-.
The dominance of the scavenging effect on possible simple processes of diffusion of the various components between contiguous snow layers seems to be proved by the asymmetry of the concentration profiles. Table 2 shows higher values for sample 6 (layer younger than sample 7) than for sample 8 (layer older than sample 7) for all the components forming soluble salts, such as Na+, CF, totS04 2-, MSA, K+, Mg2+ and Ca2+. Diffusion processes should have caused a similar increase in the contiguous layers or, better, a higher increase in the oldest layer by the longer contact time. Instead, some compounds prevalently associated with gas phase (carboxylic acids, NO3-, NH4 + and Br") show higher concentrations in sample 8 than in sample 6. This evidence could support the hypothesis of a gas emission just before the massive volcanic event.
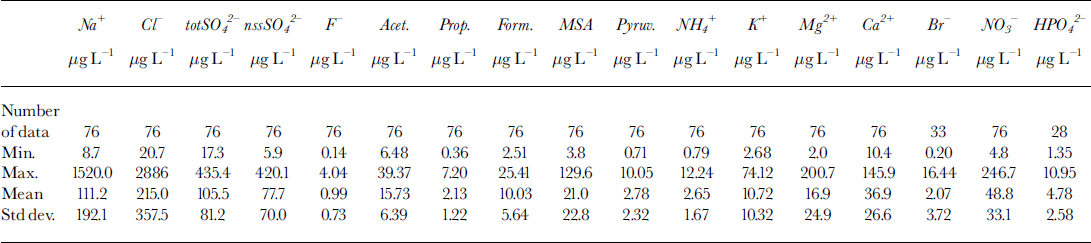
A sudden increase of concentration in sample 7 is shown by all the analyzed components, although the increase is lower than that shown by fluoride and sulphate. Figure 5 shows the concentration profiles for Ga2+ (Fig.5a), MSA (Fig.5b) and phosphate (Fig.5c). These compounds show concentration peaks in sample 7 about 30, 70 and 10 times higher, respectively, than the background values, but the concentration levels fall immediately to the background values in samples 6 and 8. This pattern is consistent with the hypothesis that the volcanic event was very time-limited and/or the source was near enough to collect the soil particles emitted in the atmosphere. In particular, Ga2+ can be used as a crustal indicator (the Ga2+/Na+ ratio in sample 7 is twice as high as in samples 6 and 8) and MSA is solely produced by biogenic marine activity, so it cannot be directly correlated to the volcanic input (ash). Phosphate, too, can be attributed to biogenic activity, but this source is not unique. Therefore, the high concentrations of calcium, MSA and phosphate in sample 7 seem to be ascribable, not to volcanic ash, but to the contribution of soil particles. In fact, the only explanation for high concentrations of MSA is gradual accumulation on the top and the slopes of the volcano by deposition and successive melting of snow precipitations. At the moment of the eruption, a quantity of the surface soil could be emitted into the atmosphere in addition to the normal ash emission. The short half-lives of these rather large soil particles caused their fast deposition, by wet or dry removal processes, so that the atmospheric scavenging was already complete at the time of the successive snow depositions (sample 6).
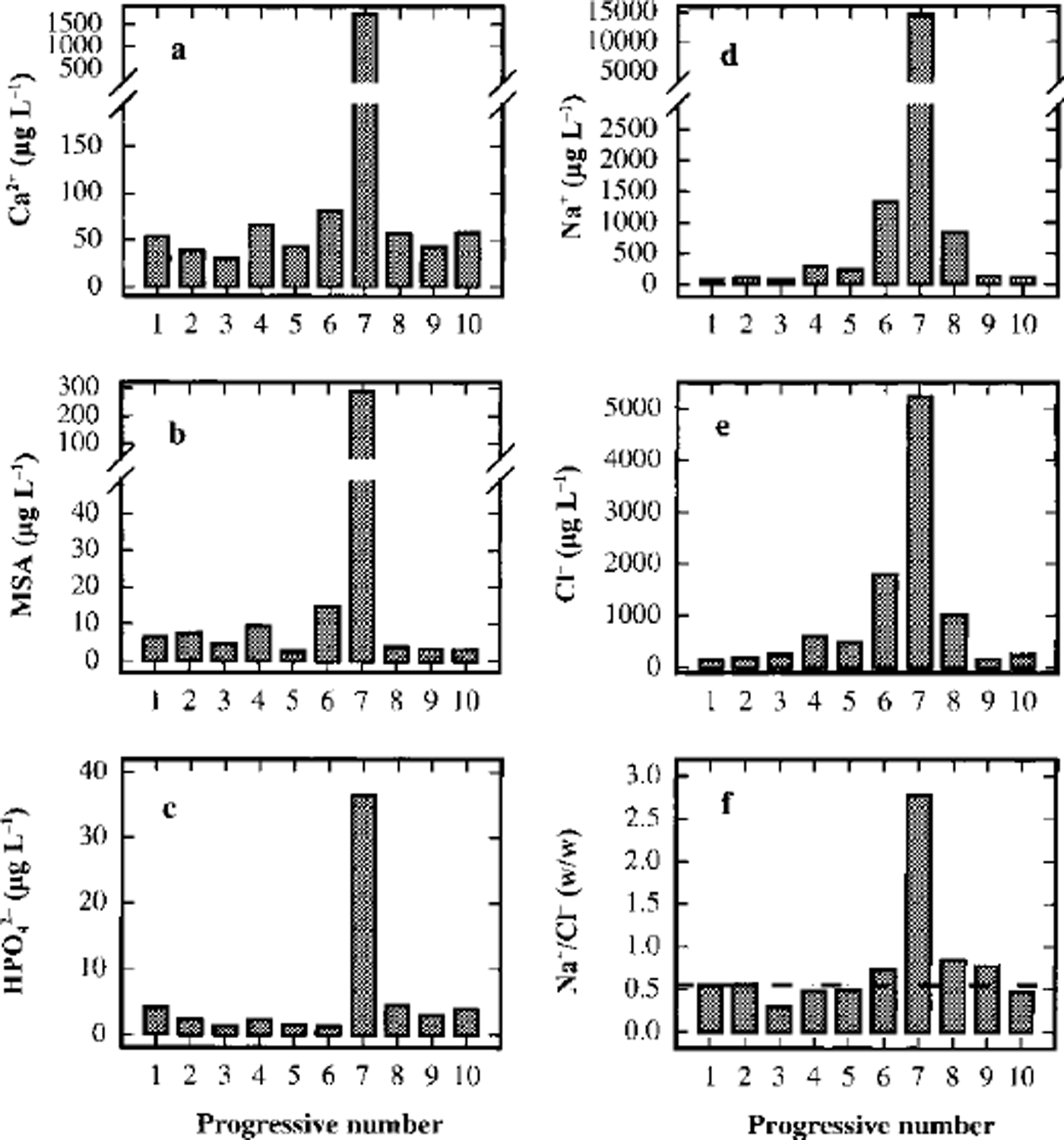
Fig. 5. Concentration/depth profiles of calcium (a), MSA (b), phosphate (c), sodium (d) and chloride (e), and trend of the Na+/Cr w/w ratio (f), in the ten subsamples around the volcanic layer from 93.23 (sample 1) to 93.50 (sample 10) m depth.
Furthermore, the MSA profile unarguably shows the absence of diffusion effects. Actually, for this compound, the only known source is biogenic, and its concentration profile cannot be misinterpreted by any secondary source or particular transport phenomena. The MSA concentration in the layers contiguous to the dark layer falls immediately to background levels (<10/μgL-1), values that are similar to the 1988-93 mean (21.0 Mg L-1; Table 3). Some authors report a seasonal dephasing between MSA and nssSO42- (with summer nssSO42- and winter MSA peaks) due to possible diffusion of MSA in recent and old snow layers in stations located in the Antarctic Peninsula (Mulvaney and others, Reference Mulvaney, Pasteur, Peel, Saltzman and Whung1992) and in the Filchner-Ronne Ice Shelf (Wagenbach and others, Reference Wagenbach1994). In our experience, this anomaly is absent from snow pits and shallow firn cores collected in northern Victoria Land (Piccardi and others, Reference Piccardi, Casella and Udisti1996b; Reference UdistiUdisti, 1996; Udisti and others, Reference Udisti, Traversi, Becagli and Piccardi1998b; Stenni and others, unpublished). The Styx Glacier medium-depth ice core confirms our previous observation that in a 500 year period no significant diffusion is shown between two contiguous snow layers, about 10 mm apart, with a concentration ratio of 80:1 (sample 7/ sample 8).
Na+ and CF (Fig.5d and e) show a lower increase in sample 7 compared with the two samples around the dark layer. The Na+ content in sample 7 is about one order of magnitude higher than in samples 6 and 8, but the CF content is only five times higher. However, the Na+ peak is about 100 times higher than the 1988-93 mean value at the same station (Table 3), so the contribution of the sea salt accumulated on the soil particles seems also to be dominant in this case. The fundamental difference between sample 7 and contiguous samples is shown in Figure 5f, which gives the Na+/Cl- ratios for the ten samples. In all other samples, the Na+/Cl- ratio is very similar to the sea-water composition (Na+/Cl- = 0.55 w/w; dashed line in Fig. 5f), so the sea-spray source seems apparent. The relatively high concentrations in samples 6 and 8, therefore, may be due to the normal variability in the sea-salt content in snow. Data from snow pits and shallow firn cores show that values higher than 1 mg L-1 for Na+ and Cl- are not unusual for this station, corresponding to snow precipitation with sea winds (Piccardi and others, Reference Piccardi, Udisiti and Casella1994a, b, 1996a). For sample 7, however, the GF/Na+ ratio is dramatically different and shifted to an Na+ preponderance (Na+/Cl- = 2.8 w/w). This pattern can be explained by the fact that following the volcanic event, the atmospheric aerosol was enriched by very high concentrations of H2SO4 and other acids. In fact, acidity measurements are a simple method of identifying volcanic layers in snow and ice (Hammer and others, Reference Hammer, Clausen and Dansgaard1980). The NaCl included in the atmospheric aerosol and on the soil particles can react with H2SO4 to give Na2SO4 and gaseous HCl (Mayewski and others, Reference Mayewski1993).
The gas-phase HCl may not be deposited in the ice, while Na2SO4 is deposited by dry and/or wet (snow) deposition. Two observations can be made:
The Na+/Cl- ratio returns quickly to sea-water values. Going to sample 6 from sample 7, the ratio changes from 2.8 to 0.74, approaching the theoretical value of 0.55.
The variation of Na+/Cl- ratio could confirm the source closeness, too, as the supposed exchange mechanism between H2SO4 and NaCl involves two processes:
-
(1) near the source: the deposition of Na2SO4 and the sending away of gaseous HCl with an increase of the Na+/CF ratio;
-
(2) far from the source: the deposition of HCl with a decrease of the Na+/Cl- ratio.
Mg2 + and K + follow the same trend as Na+, showing that sea spray is an important source, but with lower peaks in sample 7 (about 9 and 14 times the background values, respectively).
Figure 6 shows the concentration profiles for NH4 +, NO3- and BF. These components, as already mentioned, have a peculiarity: the concentration peaks in sample 7 do not show high enrichments with respect to background values (about 5, 7 and 10 times, respectively), but like F-, their concentration in sample 8 Gust before the volcanic event) is significantly higher than the background. Moreover, for NO3- and BF the concentration in sample 8 is higher than in sample 6 Gust after the dark layer) and comparable with sample 7. This is also true, but less evident, for NH4 +. A significant contribution to snow composition by dry deposition (probably snow-surface adsorption) and/or wet deposition of HNO3 and HBr (as well as F-), directly emitted or built up as secondary volcanic products, must be taken into account.
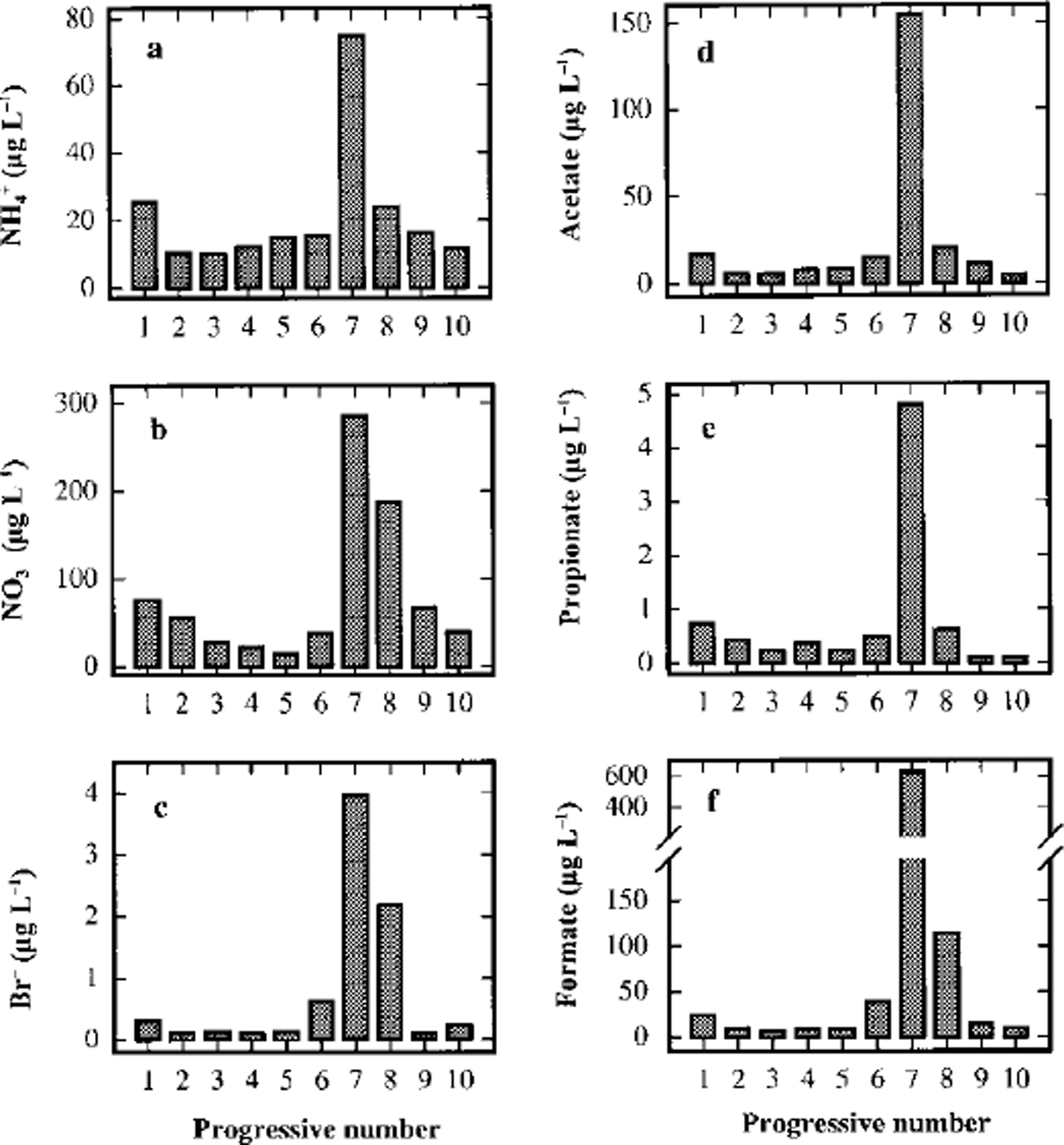
Fig. 6. Concentration/depth profiles of ammonium (a), nitrate (b), bromide (c), acetate (d), propionate (e) and formate (f) in the ten subsamples around the volcanic layer from 93.23 (sample 1) to 93.50 (sample 10) m depth.
Finally, some short-chain carboxylic acids show high concentrations in sample 7 (Fig. 6). Peak concentrations 10-20 times higher than the background values were measured for acetic, propionic and formic acids. In particular, formate has the highest concentration, reaching a value about 60 times higher than the mean 1988-93 value (10.03 μgl L-1; Table 3). The concentration of the carboxylic acids, too, are higher in sample 6 than in sample 8, confirming the similar pattern of atmospheric gas-phase distribution compounds.
Many sources were postulated for carboxylic acids in polar atmosphere, such as direct input from biologic activity and vegetation, secondary emission by oxidation of marine and continental biogenic hydrocarbons or biomass-burning, and from anthropogenic activity (Reference Talbot, Andreae, Berresheim, Jacob and BeecherTalbot and others, 1990; Legrand and De Angelis, 1996; Reference Udisti, Becagli, Traversi, Vermigli and PiccardiUdisti and others, 1998a). Measurements of acetic, propionic and formic acids in polar snow are sporadic, and no correlation was reported between their increase and the volcanic activity Reference Legrand and de AngelisLegrand and De Angelis (1996) report an inverse correlation between carboxylic- acid concentrations in the snow and high acidic content in the atmosphere such as happens during a volcanic eruption. Even so, in Antarctica their production is mainly related to carbon-cycle oxidation processes, so they could also be built up by oxidation of hydrocarbons emitted by volcanic activity. For example, Reference BrimblecombeBrimblecombe (1996) reports a CH4 global flux of 0.34 Tg a-1 from volcanic input. When the carboxylic-acid measurements are available for Antarctic ice cores, where anthropogenic, biomass-burning and vegetation emissions are very small, the correlation with volcanic activity will be verified.
What is the source?
A comparison with the explosive volcanic records at South Pole in the last 1000 years (Reference Delmas, Kirchner, Palais and PetitDelmas and others, 1992) did not give unambiguous information to identify the source. No eruptions were reported for the period around AD 1500. The Ruiz and Huaynaputina eruptions (dating of the signal: AD 1596 and 1601, respectively) have a large time difference from the Styx event dating (about 50 years). Besides, their sulphate signals in the ice are not as large as that found in the Styx Glacier volcanic layer. A high sulphate peak characterises the closer eruption around AD 1450. Reference Delmas, Kirchner, Palais and PetitDelmas and others (1992) suggest that Deception Island or Sandwich Island could be the source. Reference ZielinskiZielinski (1995) attributes the 1450 volcanic signal to the Kuwae eruption (GISP2 Greenland ice-core analysis). A better comparison will be carried out once a more reliable dating of Styx Glacier ice core is available by chemical and isotopic «5180 profiles (now in progress), and using time-reference horizons.
The thickness of the dark layer and, above all, the fast decrease of all the marker concentrations within a very short distance of the zone with the highest values suggest that this is a local volcanic event. Besides, a contribution of dry deposition of particulate material containing relatively large soil particles is evident, in addition to the ash deposition. This is shown by high concentrations of typical crustal substances such as Ga2+, Mg2 + and K+ and of other compounds that cannot be related to volcanic ash but only to soil-accumulation processes by snow-melting, such as MSA and phosphate. These relatively large soil particles can reach the deposition site by two principal processes: fast dry deposition if the source is near, or injection into the high troposphere (or into the stratosphere) in the case of explosive volcanic events for very distant sources. In the second case, however, a longer half-life in the atmosphere is predictable, with the removal processes (by dry and wet depositions) continuing for a long time, leading to the markers being recorded in a thicker ice-core layer.
The Styx Glacier station is located about 40 km from the Mount Melbourne volcano, the eruptive activity of which is described by Reference Worner, Niephaus, Hertogen and ViereckWorner and others (1989), Reference ArmientiArmienti and others (1991), Reference Horning, Worner and ZipfelHorning and others (1991) and Reference Lanzafame and VillariLanzafame and Villari (1991). Structural and chemical analysis of the ice-core particulate compared to Mount Melbourne eruption deposits will clarify whether this is the source.
A more probable source is Mount Erebus, a still fully active volcano about 300 km from Styx Glacier. Reference Zreda-Gostynska, Kyle, Finnegan and PrestboZreda-Gostynska and others (1997) report high concentrations of HF in gas and aerosol measurements of Mount Erebus emissions, and support F- concentration in snow being a tracer for Erebus volcanic activity. Reference Zreda-Gostynska, Kyle, Finnegan and PrestboZreda-Gostynska and others (1997)estimate an HF emission rate of 2.3-9.2 Gg a-1 in the period December 1986-January 1991, with an air concentration of 2.5-5 ng m-3 and a residence time of 10-21 days. Mount Erebus also emits H2SO4 (25.9 ± 7.3 Gga-1 as SO2 in January 1991), but this contribution is less distinctive and comprises only a small percentage (about 3%) of the total sulphur budget in the Antarctic atmosphere.
Reference Aristarain and DelmasAristarain and Delmas (1998) report a well-marked volcanic ash layer in a 145.9 m ice core from James Ross Island and attribute it to Deception Island volcano (around AD 1641). A comparison between the chemical markers reported by Aristarain and Delmas and our data seems unsatisfactory. The time shift is too large, even considering that our dating is not definitive. Similar CF depletion, with respect to the sea-water Na+/CF ratio, is reported, but no increase of nitrate is shown in the Deception Island eruption, and the sulphate increase is much smaller (only about 4.5 times higher than the background level), even if the ice-core station (James Ross Island) is rather close to (about 200 km from) the source. For the Styx Glacier ice core, located about 4500 km from Deception Island, sulphate concentration in the dark layer is 370 times higher than the background level. Aristarain and Delmas report no fluoride data. They also report the chronological scale of the Deception Island eruptions in the last millennium, indicating a volcanic event dated around AD 1550 identified by marine and lake sediment (Fildes Peninsula-Bransfield Strait Area). Unfortunately, the James Ross ice core is too short to characterise such an eruption in the snow.
Volcanoes located outside Antarctica or even in the Northern Hemisphere seem to be less probable sources. Long-range transport processes are highly dependent on the event intensity and meteorological conditions, such as seasonality and strength and direction of the winds. Measurements performed on various transport tracers (Reference Wagenbach, Wolff and BalesWagenbach, 1996) showed that the characteristic time for the inter-hemispheric exchange is around lyear. This is more than an order of magnitude greater than the typical tropo-spheric aerosol residence time. Therefore, only cataclysmic events of global concern can be recorded in snow and ice cores. In the Styx volcanic layer, a cataclysmic explosive eruption can be excluded, since it would be memorized in all Antarctic ice cores due to the very high-concentrate deposits, and only a regional input seems probable. Therefore, we conclude that the source is located in the Southern Hemisphere, probably on the Antarctic continent.
Conclusions
A roughly 500 year-old volcanic event was identified and characterised by chemical analysis of a 110 m ice core drilled at Styx Glacier plateau.
The event was characterised by a gas emission (HF, SO2 and probably HBr, HNO3 and NH3) followed by a volcanic eruption with emission of ash and probably soil particulate into the atmosphere. Gas-phase and particulate atmospheric scavenging occurred by dry and wet removal processes in a very short time (about 10 mm ice layer in a station with a mean snow accumulation rate of about 160 kg m-2 a-1; Reference Piccardi, Udisiti and CasellaPiccardi and others, 1994a; Reference UdistiUdisti, 1996; Stenni and others, unpublished). The most obvious markers were HF and H2SO4 for gas-phase emission and Ga2+ and MSA (indirect indicators) for soil-particulate scavenging. Significant concentrations of nitrate, ammonium and bromide are present in and directly beneath the dark layer, indicating a possible early emission as gas-phase compounds. The possibility that high concentrations of some carboxylic acids (acetic, propionic and formic) can constitute a volcanic marker needs to be confirmed.
The acidic emission (primarily H2SO4) probably caused a sharp increase of Na+/Cl- ratio with respect to the sea-water composition by HCl removal.
No significant diffusion between adjacent snow layers (resolution 30 mm) was shown by the concentration/depth profiles.
A source attribution of the Styx Glacier volcanic event is not yet possible. The most likely candidate is Mount Erebus, in view of its massive fluoride emission and its proximity The chemical and structural analysis of the particulate, now in progress, will clarify the problem.
Acknowledgements
This research was carried out in the framework of a project on glaciology and palaeoclimatology of the Programma Nazionale di Ricerche in Antartide. It was financially supported by Ente per le Nuove Tecnologie, l’Energia e l’Ambiente through a cooperative agreement with Università di Milano.









