
Recyclable Ir Nanoparticles for the Catalytic Hydrogenation of Biomass-Derived Carbonyl Compounds

 , , , and
, , , and
Abstract
:
1. Introduction
2. Results and Discussion
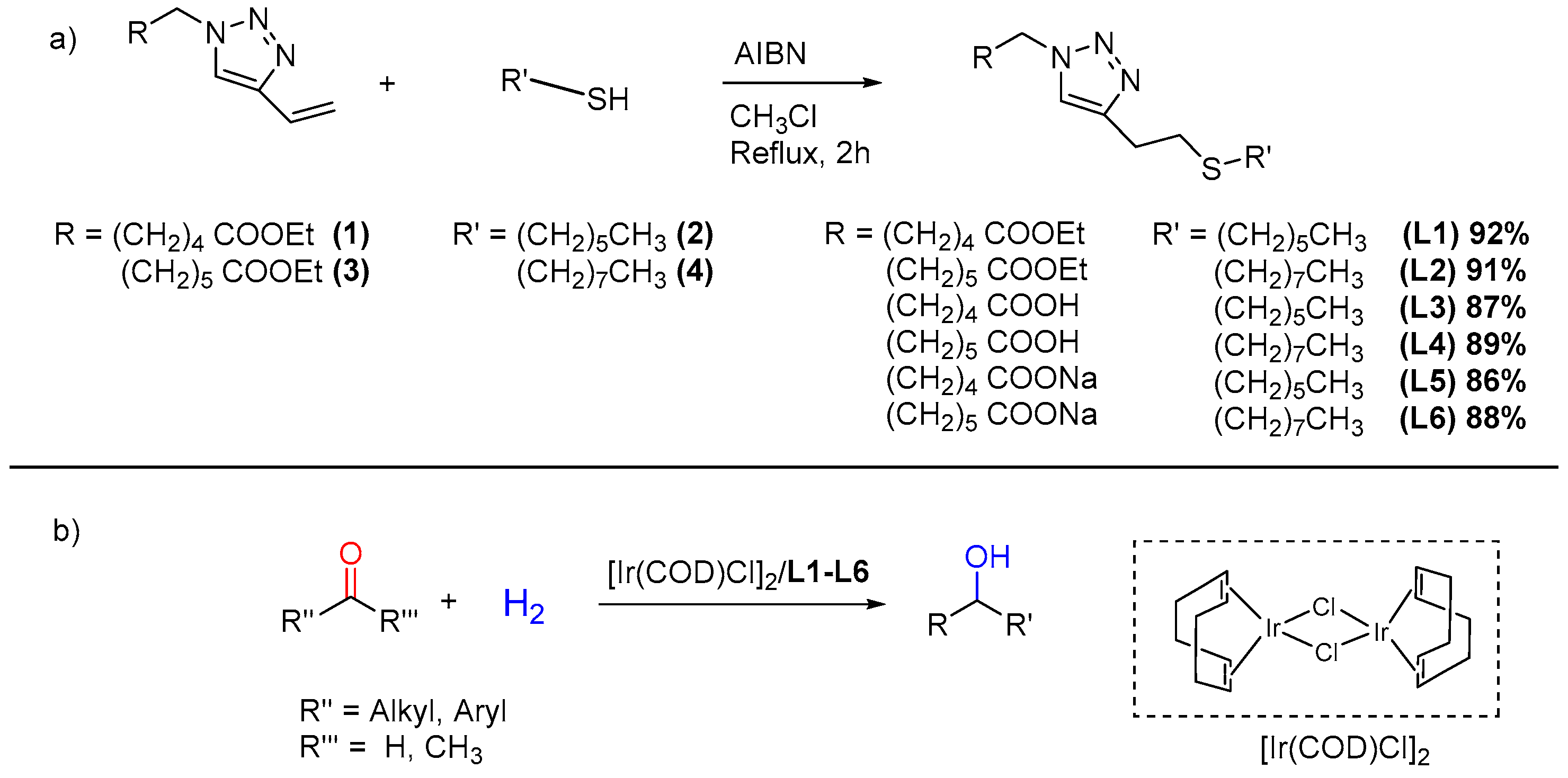
2.1. Ligand Synthesis
2.2. Hydrogenation of Furfural
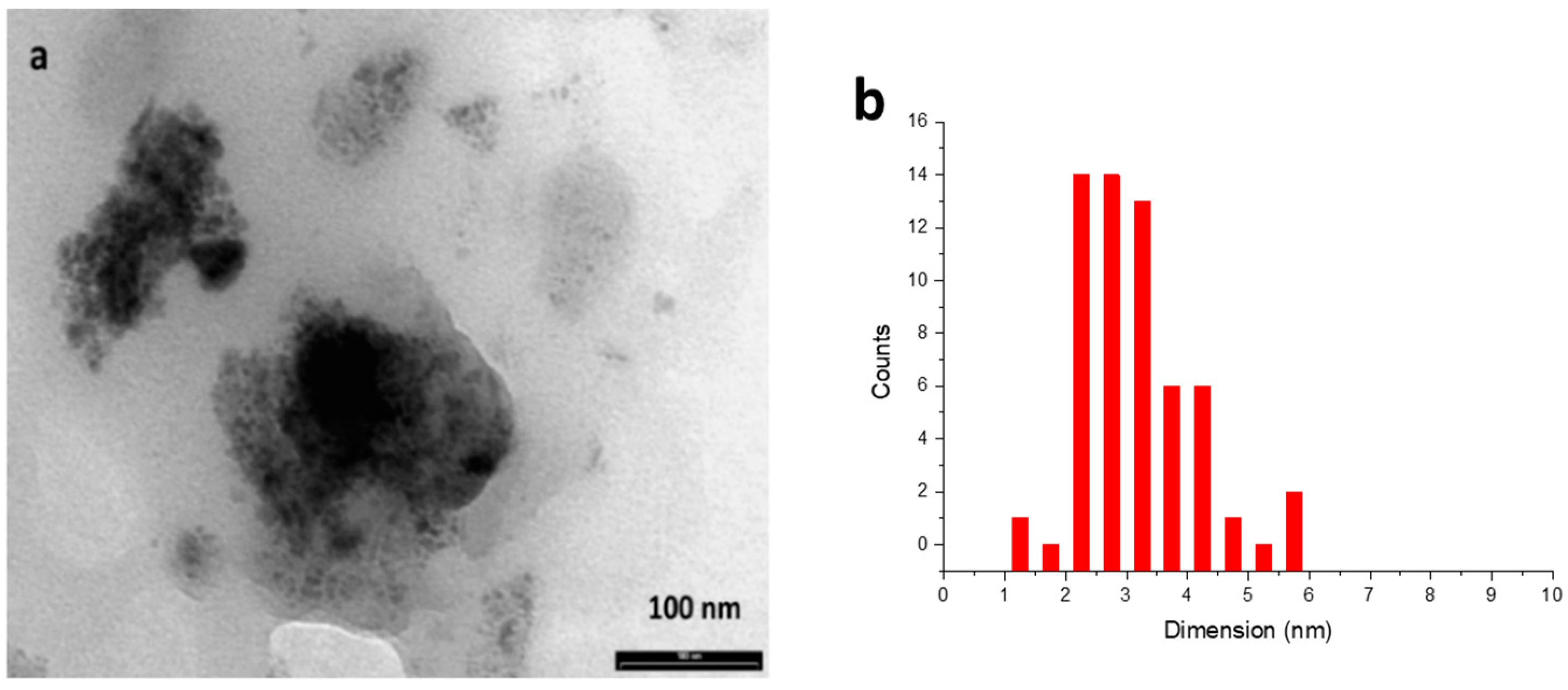
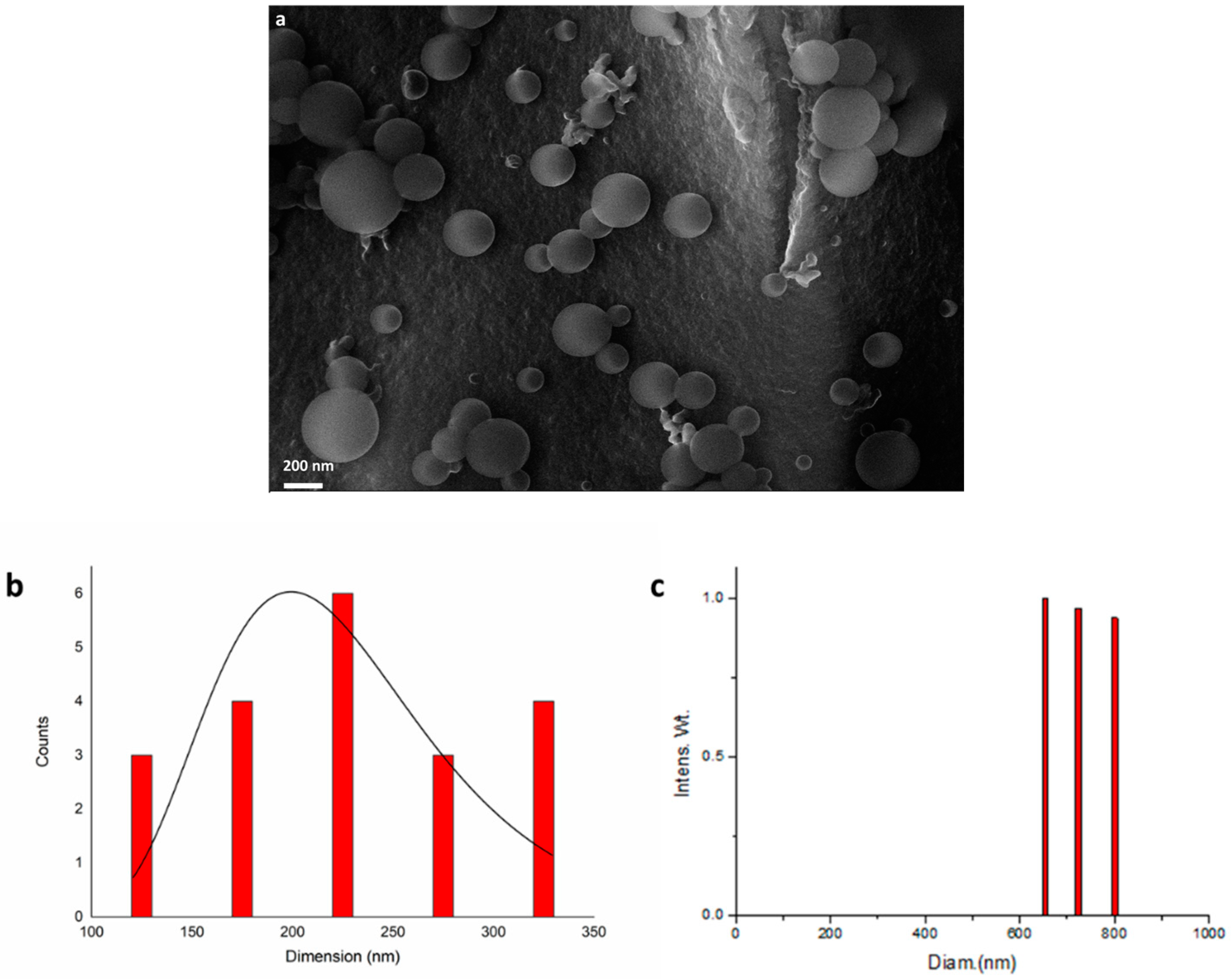
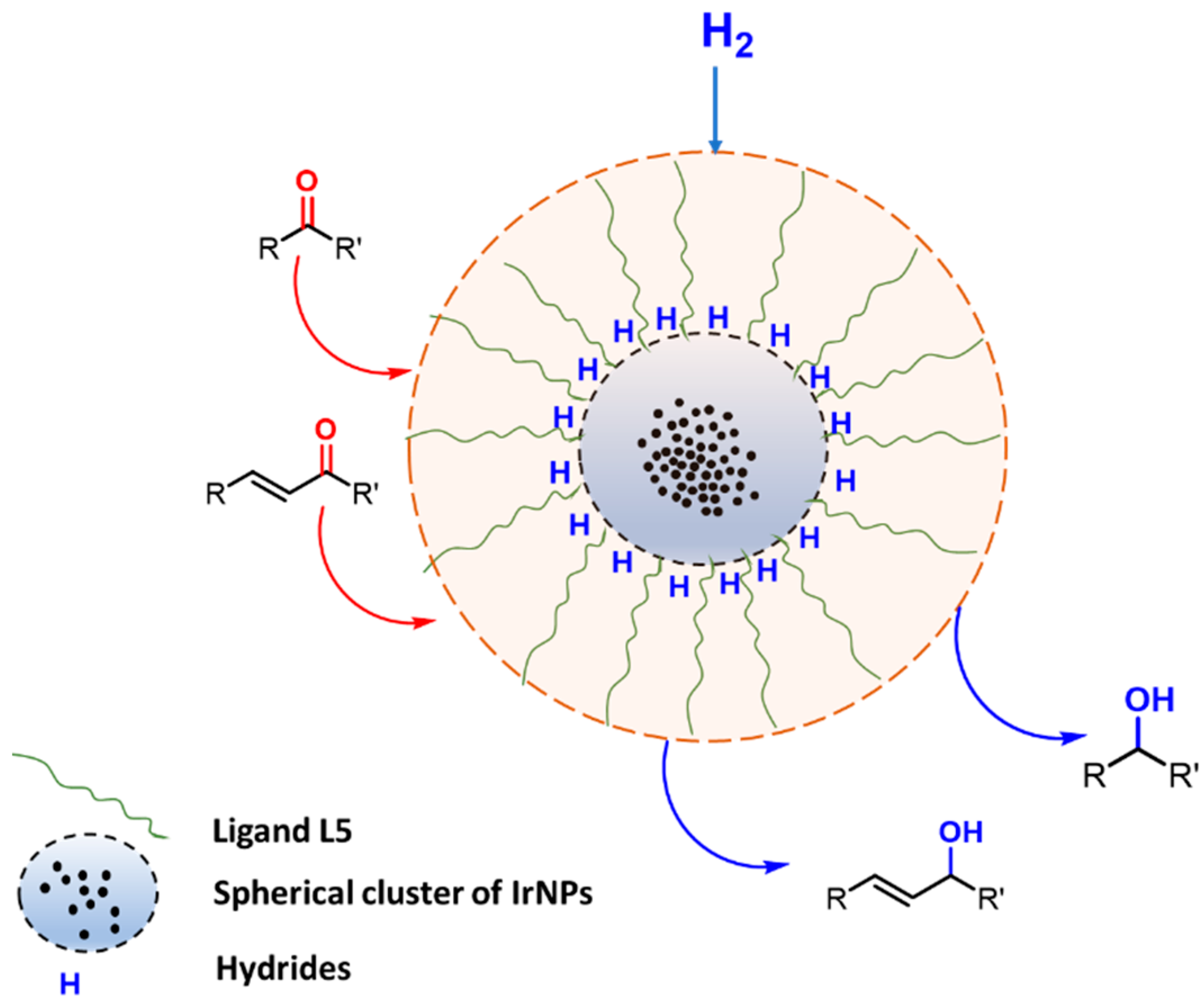
2.3. Characterisation of IrNPs
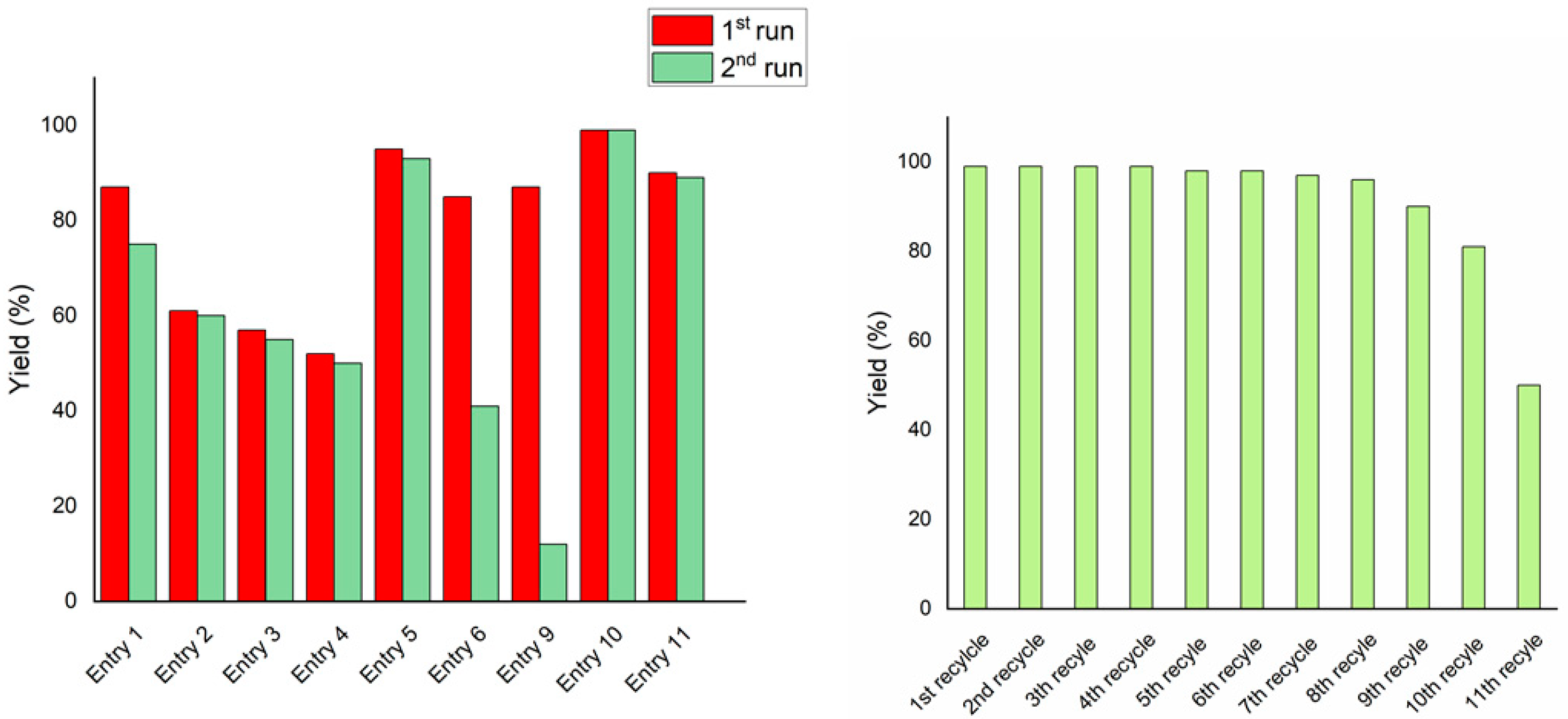
2.4. Hydrogenation of Biomass-Derived Platform Chenicals
3. Materials and Methods
3.1. Materials
3.2. Instrumentation
3.3. Generic Procedure for Catalytic Hydrogenation
3.4. Ligand Synthesis
3.4.1. Synthesis of Ethyl 5-(4-(2-(hexylthio)ethyl)-1H-1,2,3-triazol-1-yl)pentanoate (L1)
3.4.2. Synthesis of Ethyl 5-(4-(2-(hexylthio)ethyl)-1H-1,2,3-triazol-1-yl)hexanoate (L2)
3.4.3. Synthesis of 5-(4-(2-(hexylthio)ethyl)-1H-1,2,3-triazol-1-yl)pentanoic Acid (L3)
3.4.4. Synthesis of 5-(4-(2-(hexylthio)ethyl)-1H-1,2,3-triazol-1-yl)hexanoicanoic Acid (L4)
3.4.5. Synthesis of Sodium 5-(4-(2-(hexylthio)ethyl)-1H-1,2,3-triazol-1-yl)pentanoate (L5)
3.4.6. Synthesis of Sodium 5-(4-(2-(hexylthio)ethyl)-1H-1,2,3-triazol-1-yl)pentanoate (L6)
3.5. Product Characterisation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Cornils, B.; Herrmann, W.A.; Beller, M.; Paciello, R. (Eds.) Applied Homogeneous Catalysis with Organometallic Compounds; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2017; ISBN 9788578110796. [Google Scholar]
- De Vries, J.G.; Elsevier, C.J. (Eds.) The Handbook of Homogeneous Hydrogenation; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006; ISBN 9783527311613. [Google Scholar]
- Li, A.Y.; Moores, A. Carbonyl Reduction and Biomass: A Case Study of Sustainable Catalysis. ACS Sustain. Chem. Eng. 2019, 7, 10182–10197. [Google Scholar] [CrossRef]
- Paganelli, S.; Alam, M.M.; Beghetto, V.; Scrivanti, A.; Amadio, E.; Bertoldini, M.; Matteoli, U. A pyridyl-triazole ligand for ruthenium and iridium catalyzed C=C and C=O hydrogenations in water/organic solvent biphasic systems. Appl. Catal. A Gen. 2015, 503, 20–25. [Google Scholar] [CrossRef]
- Anastas, P.; Eghbali, N. Green Chemistry: Principles and Practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, R. Catalytic Hydrogenation for Biomass Valorization; Rinaldi, R., Ed.; Royal Society of Chemistry: Cambridge, UK, 2015; ISBN 9781782620099. [Google Scholar]
- Sole, R.; Taddei, L.; Franceschi, C.; Beghetto, V. Efficient Chemo-Enzymatic Transformation of Animal Biomass Waste for Eco-Friendly Leather Production. Molecules 2019, 24, 2979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornils, B.; Herrmann, W.A.; Horvat, J.; Leitner, W.; Mecking, S.; Olivier-Bourbigou, H.; Vogt, D. (Eds.) Multiphase Homogenous Catalysis; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005; ISBN 9783527307210. [Google Scholar]
- Scrivanti, A.; Bortoluzzi, M.; Sole, R.; Beghetto, V. Synthesis and characterization of yttrium, europium, terbium and dysprosium complexes containing a novel type of triazolyl–oxazoline ligand. Chem. Pap. 2018, 72, 799–808. [Google Scholar] [CrossRef]
- Cornils, B.; Herrmann, W.A. Aqueous-Phase Organometallic Catalysis; Cornils, B., Herrmann, W.A., Eds.; Wiley-VCH Verlag: Weinheim, Germany, 2004; ISBN 3527307125. [Google Scholar]
- Beghetto, V.; Scrivanti, A.; Bertoldini, M.; Aversa, M.; Zancanaro, A.; Matteoli, U. A practical, enantioselective synthesis of the fragrances canthoxal and silvial®, and evaluation of their olfactory activity. Synthesis 2015, 47, 272–278. [Google Scholar] [CrossRef]
- Kliewer, C.J.; Bieri, M.; Somorjai, G.A. Hydrogenation of the α,β-unsaturated aldehydes acrolein, crotonaldehyde, and prenal over Pt single crystals: A kinetic and sum-frequency generation vibrational spectroscopy study. J. Am. Chem. Soc. 2009, 131, 9958–9966. [Google Scholar] [CrossRef] [PubMed]
- Astruc, D. (Ed.) Nanoparticles and Catalysis; Wiley-VCH Verlag: Weinheim, Germany, 2008; ISBN 9783527305070. [Google Scholar]
- Serp, P.; Philippot, K. (Eds.) Nanomaterials in Catalysis: First Edition; Wiley-VCH Verlag: Weinheim, Germany, 2012; ISBN 9783527331246. [Google Scholar]
- Martínez-Prieto, L.M.; Chaudret, B. Organometallic Ruthenium Nanoparticles: Synthesis, Surface Chemistry, and Insights into Ligand Coordination. Acc. Chem. Res 2018, 51, 376–384. [Google Scholar] [CrossRef]
- Ruiz-Varilla, A.M.; Baquero, E.A.; Chaudret, B.; de Jesús, E.; Gonzalez-Arellano, C.; Flores, J.C. Water-soluble NHC-stabilized platinum nanoparticles as recoverable catalysts for hydrogenation in water. Catal. Sci. Technol. 2020, 10, 2874–2881. [Google Scholar] [CrossRef]
- Li, W.; Wang, Y.; Chen, P.; Zeng, M.; Jiang, J.; Jin, Z. Thermoregulated phase-transfer iridium nanoparticle catalyst: Highly selective hydrogenation of the C=O bond for α,β-unsaturated aldehydes and the C=C bond for α,β-unsaturated ketones. Catal. Sci. Technol. 2016, 6, 7386–7390. [Google Scholar] [CrossRef]
- Cano, I.; Tschan, M.J.L.; Martínez-Prieto, L.M.; Philippot, K.; Chaudret, B.; Van Leeuwen, P.W.N.M. Enantioselective hydrogenation of ketones by iridium nanoparticles ligated with chiral secondary phosphine oxides. Catal. Sci. Technol. 2016, 6, 3758–3766. [Google Scholar] [CrossRef]
- Kolb, H.C.; Sharpless, K.B. The growing impact of click chemistry on drug discovery. Drug Discov. Today 2003, 8, 1128–1137. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Beghetto, V.; Gatto, V.; Conca, S.; Bardella, N.; Buranello, C.; Gasparetto, G.; Sole, R. Development of 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium chloride cross-linked carboxymethyl cellulose films. Carbohydr. Polym. 2020, 249, 116810. [Google Scholar] [CrossRef]
- Sole, R.; Agostinis, L.; Conca, S.; Gatto, V.; Bardella, N.; Morandini, A.; Buranello, C.; Beghetto, V. Synthesis of Amidation Agents and Their Reactivity in Condensation Reactions. Synthesis 2021, 53, 1672–1682. [Google Scholar] [CrossRef]
- Sole, R.; Gatto, V.; Conca, S.; Bardella, N.; Morandini, A.; Beghetto, V. Sustainable Triazine-Based Dehydro-Condensation Agents for Amide Synthesis. Molecules 2021, 26, 191. [Google Scholar] [CrossRef]
- Hoyle, C.E.; Lowe, A.B.; Bowman, C.N. Thiol-click chemistry: A multifaceted toolbox for small molecule and polymer synthesis. Chem. Soc. Rev. 2010, 39, 1355–1387. [Google Scholar] [CrossRef]
- Hoyle, C.E.; Bowman, C.N. Polymer Chemistry Thiol—Ene Click Chemistry. Angew. Chem. Int. Ed. 2010, 49, 1540–1573. [Google Scholar] [CrossRef]
- Huang, S.; Sinha, J.; Podgo, M.; Zhang, X.; Claudino, M.; Bowman, C.N. Mechanistic Modeling of the Thiol−Michael Addition Polymerization Kinetics: Structural Effects of the Thiol and Vinyl Monomers. Macromolecules 2018, 51, 5979–5988. [Google Scholar] [CrossRef]
- Nair, D.P.; Podgórski, M.P.; Chatani, S.; Gong, T.; Xi, W.; Fenoli, C.R.; Bowman, C.N. The Thiol-Michael Addition Click Reaction: A Powerful and Widely Used Tool in Materials Chemistry. Chem. Mater. 2013, 26, 724–744. [Google Scholar] [CrossRef]
- Takizawa, K.; Nulwala, H.; Thibault, R.J.; Lowenhielm, P.; Yoshinaga, K.; Wooley, K.L.; Hawker, C.J. Facile Syntheses of 4-Vinyl-1,2,3-triazole Monomers by Click Azide/Acetylene Coupling. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 2897–2912. [Google Scholar] [CrossRef]
- Mariscal, R.; Maireles-Torres, P.; Ojeda, M.; Sádaba, I.; López Granados, M. Furfural: A renewable and versatile platform molecule for the synthesis of chemicals and fuels. Energy Environ. Sci. 2016, 9, 1144–1189. [Google Scholar] [CrossRef]
- Deuss, P.J.; Barta, K.; De Vries, J.G. Homogeneous catalysis for the conversion of biomass and biomass-derived platform chemicals. Catal. Sci. Technol. 2014, 4, 1174–1196. [Google Scholar] [CrossRef] [Green Version]
- Puylaert, P.; van Heck, R.; Fan, Y.; Spannenberg, A.; Baumann, W.; Beller, M.; Medlock, J.; Bonrath, W.; Lefort, L.; Hinze, S.; et al. Selective Hydrogenation of α,β-Unsaturated Aldehydes and Ketones by Air-Stable Ruthenium NNS Complexes. Chemistry 2017, 23, 8473–8481. [Google Scholar] [CrossRef]
- Werkmeister, S.; Neumann, J.; Junge, K.; Beller, M. Pincer-Type Complexes for Catalytic (De)Hydrogenation and Transfer (De)Hydrogenation Reactions: Recent Progress. Chemistry 2015, 21, 12226–12250. [Google Scholar] [CrossRef] [PubMed]
- Scrivanti, A.; Beghetto, V.; Alam, M.M.; Paganelli, S.; Canton, P.; Bertoldini, M.; Amadio, E. Biphase hydroformylation catalyzed by rhodium in combination with a water-soluble pyridyl-triazole ligand. Inorg. Chim. Acta 2017, 455, 613–617. [Google Scholar] [CrossRef]
- Li, J.; Li, X.; Ma, Y.; Wu, J.; Wang, F.; Xiang, J.; Zhu, J.; Wang, Q.; Deng, J. Surfactant-accelerated asymmetric transfer hydrogenation with recyclable water-soluble catalyst in. RSC Adv. 2013, 3, 1825–1834. [Google Scholar] [CrossRef]
- Jansat, S.; Picurelli, D.; Pelzer, K.; Philippot, K.; Gómez, M.; Muller, G.; Lecante, P.; Chaudret, B. Synthesis, characterization and catalytic reactivity of ruthenium nanoparticles stabilized by chiral N-donor ligands. New J. Chem. 2006, 30, 115–122. [Google Scholar] [CrossRef]
- An, K.; Somorjai, G.A. Size and Shape Control of Metal Nanoparticles for Reaction Selectivity in Catalysis. ChemCatChem 2012, 4, 1512–1524. [Google Scholar] [CrossRef]
- Li, J.; Tang, Y.; Wang, Q.; Li, X.; Cun, L.; Zhang, X.; Zhu, J.; Li, L.; Deng, J. Chiral Surfactant-Type Catalyst for Asymmetric Reduction of Aliphatic Ketones in Water. J. Am. Chem. Soc. 2012, 134, 18522–18525. [Google Scholar] [CrossRef]
- Lipshutz, B.H.; Lipshutz, B.H.; Isley, N.A.; Fennewald, J.C.; Slack, E.D. On the Way Towards Greener Transition-Metal- Catalyzed Processes as Quantified by E Factors. Angew. Chem. Int. Ed. 2013, 52, 10952–10958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Sorella, G.; Strukul, G.; Scarso, A. Recent advances in catalysis in micellar media. Green Chem. 2015, 17, 644. [Google Scholar] [CrossRef]
- Pery, T.; Pelzer, K.; Buntkowsky, G.; Philippot, K.; Limbach, H.-H.; Chaudret, B. Direct NMR Evidence for the Presence of Mobile Surface Hydrides on Ruthenium Nanoparticles. ChemPhysChem 2005, 6, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Bozell, J.J.; Petersen, G.R. Technology development for the production of biobased products from biorefinery carbohydrates—The US Department of Energy’s “Top 10” revisited. Green Chem. 2010, 12, 539–554. [Google Scholar] [CrossRef]
- Program, B.; Werpy, T.; Petersen, G. Top Value Added Chemicals from Biomass Volume I-Results of Screening for Potential Candidates from Sugars and Synthesis Gas; Pacific Northwest National Laboratory (PNNL): Richland, WA, USA; National Renewable Energy Laboratory (NREL) Office of Biomass: Golden, CO, USA, 2004. [Google Scholar]
- Sun, Z.; Fridrich, B.; De Santi, A.; Elangovan, S.; Barta, K. Bright Side of Lignin Depolymerization: Toward New Platform Chemicals. Chem. Rev. 2018, 118, 614–678. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Xu, J.; Zhou, S.; He, A.; Tang, X.; Lin, L.; Xu, J.; Zhao, Y. Catalytic Advances in the Production and Application of Biomass-Derived 2,5-Dihydroxymethylfuran. ACS Catal. 2018, 8, 2959–2980. [Google Scholar] [CrossRef]
- Sole, R.; Bortoluzzi, M.; Spannenberg, A.; Tin, S.; Beghetto, V.; de Vries, J.G. Synthesis, characterization and catalytic activity of novel ruthenium complexes bearing NNN click based ligands. Dalt. Trans. 2019, 48, 13580–13588. [Google Scholar] [CrossRef]
- Van Putten, R.J.; Van Der Waal, J.C.; De Jong, E.; Rasrendra, C.B.; Heeres, H.J.; De Vries, J.G. Hydroxymethylfurfural, a versatile platform chemical made from renewable resources. Chem. Rev. 2013, 113, 1499–1597. [Google Scholar] [CrossRef] [PubMed]
- Stolle, A.; Gallert, T.; Schmöger, C.; Ondruschka, B. Hydrogenation of citral: A wide-spread model reaction for selective reduction of α,β-unsaturated aldehydes. RSC Adv. 2013, 3, 2112–2153. [Google Scholar] [CrossRef]
- Armarego, W.L.F. (Ed.) Purification of Laboratory Chemicals, 8th ed.; Elsevier Science Publishers B.V.: Amsterdam, The Netehrlands, 2017. [Google Scholar]
- Bellè, A.; Tabanelli, T.; Fiorani, G.; Perosa, A.; Cavani, F.; Selva, M. A Multiphase Protocol for Selective Hydrogenation and Reductive Amination of Levulinic Acid with Integrated Catalyst Recovery. ChemSusChem 2019, 12, 3343–3354. [Google Scholar] [CrossRef] [PubMed]
- Figliolia, R.; Cavigli, P.; Comuzzi, C.; Del Zotto, A.; Lovison, D.; Strazzolini, P.; Susmel, S.; Zuccaccia, D.; Ballico, M.; Baratta, W. CNN pincer ruthenium complexes for efficient transfer hydrogenation of biomass-derived carbonyl compounds. Dalt. Trans. 2020, 49, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Wajs-Bonikowska, A.; Malarz, J.; Stojakowska, A. Composition of Essential Oils from Roots and Aerial Parts of Carpesium divaricatum, a Traditional Herbal Medicine and Wild Edible Plant from South-East Asia, Grown in Poland. Molecules 2019, 24, 4418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, M.; Liu, Q.Z.; Liu, Z.L. Identification of insecticidal constituents from the essential oil from the aerial parts Stachys riederi var. Japonica. Molecules 2018, 23, 1200. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||
| Entry a | Ligand | Yield (%) b |
| 1 | / | 46 c |
| 2 d | / | n.d. |
| 3 | L1 | 23 |
| 4 | L2 | 31 |
| 5 | L3 | 11 |
| 6 | L4 | 5 |
| 7 | L5 | 83 |
| 8 d | L5 | 80 |
| 9 | L6 | 65 |
| 10 e | L5 | 54 |
| 11 f | L5 | 68 |
| 12 g | L5 | 70 |
 | |||||
| Entry a | t (h) | T (°C) | NaOH (eq.) b | Ir/L5 Ratio | Yield (%) c |
| 1 | 18 | 60 | 10 | 1:0.5 | 87 |
| 2 | 18 | 80 | 10 | 1:2 | 61 |
| 3 | 18 | 80 | 10 | 1:1 | 57 |
| 4 | 18 | 80 | 10 | 1:0.75 | 73 |
| 5 | 18 | 80 | 10 | 1:0.5 | 95 |
| 6 | 18 | 80 | 10 | 1:0.25 | 85 |
| 7 d | 18 | 80 | 10 | 1:0.5 | 84 |
| 8 | 8 | 80 | 10 | 1:0.5 | 66 |
| 9 | 18 | 80 | 1 | 1:0.5 | 87 |
| 10 | 18 | 80 | 2 | 1:0.5 | >99 (98 e) |
| 11 | 18 | 80 | >10 | 1:0.5 | 90 |
 | ||||
| Entry a | Substrate | Product | Yield (%) b | Recycling |
| 1 |  |  | >99 | >99 |
| 2 c |  |  | 92 | 91 |
| 3 |  |  | >99 | >99 |
| 4 |  |  | 58 | 55 |
| 5 |  |  | 88 | 86 |
| 6 |  |  | 75 | 74 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sole, R.; Buranello, C.; Bardella, N.; Di Michele, A.; Paganelli, S.; Beghetto, V. Recyclable Ir Nanoparticles for the Catalytic Hydrogenation of Biomass-Derived Carbonyl Compounds. Catalysts 2021, 11, 914. https://doi.org/10.3390/catal11080914
Sole R, Buranello C, Bardella N, Di Michele A, Paganelli S, Beghetto V. Recyclable Ir Nanoparticles for the Catalytic Hydrogenation of Biomass-Derived Carbonyl Compounds. Catalysts. 2021; 11(8):914. https://doi.org/10.3390/catal11080914
Chicago/Turabian StyleSole, Roberto, Chiara Buranello, Noemi Bardella, Alessandro Di Michele, Stefano Paganelli, and Valentina Beghetto. 2021. "Recyclable Ir Nanoparticles for the Catalytic Hydrogenation of Biomass-Derived Carbonyl Compounds" Catalysts 11, no. 8: 914. https://doi.org/10.3390/catal11080914