Looking for the “Dream Catalyst” for Hydrogen Peroxide Production from Hydrogen and Oxygen



1
CATMAT Lab, Department of Molecular Sciences and Nanosystems, Ca’ Foscari University of Venice and INSTM RUVe, via Torino 155, 30172 Venezia Mestre, Italy
2
Department of Molecular Sciences and Nanosystems, Ca’ Foscari University of Venice, Via Torino 155, 30172 Venezia Mestre, Italy
*
Authors to whom correspondence should be addressed.
Catalysts 2019, 9(3), 251; https://doi.org/10.3390/catal9030251
Submission received: 16 January 2019
/
Revised: 28 February 2019
/
Accepted: 4 March 2019
/
Published: 11 March 2019
(This article belongs to the Special Issue Direct Synthesis of Hydrogen Peroxide)
Abstract
:The reaction between hydrogen and oxygen is in principle the simplest method to form hydrogen peroxide, but it is still a “dream process”, thus needing a “dream catalyst”. The aim of this review is to analyze critically the different heterogeneous catalysts used for the direct synthesis of H2O2 trying to determine the features that the ideal or “dream catalyst” should possess. This analysis will refer specifically to the following points: (i) the choice of the metal; (ii) the metal promoters used to improve the activity and/or the selectivity; (iii) the role of different supports and their acidic properties; (iv) the addition of halide promoters to inhibit undesired side reactions; (v) the addition of other promoters; (vi) the effects of particle morphology; and (vii) the effects of different synthetic methods on catalyst morphology and performance.
1. Introduction
Hydrogen peroxide (H2O2) is considered a green oxidant and has been listed as one of the 100 most important chemicals in the world [1]. However, the method by which it is manufactured is far from being considered a sustainable one. The process via anthraquinone autoxidation is now over 70 years old but still the only one commercially used to produce H2O2, even if it suffers from many drawbacks including the disposal of considerable amounts of toxic organic wastes and the need to be run in large plants to compensate for the high operating costs. Therefore, despite its advantages as a clean oxidant, H2O2 is not produced via an environmentally friendly process and is still rather costly (current price is about 500 US $ per ton for 50% hydrogen peroxide [2]), thus not economically competitive for the production of bulk chemicals or for a more widespread use in wastewater treatment. For these reasons, the development of a green process for the synthesis of H2O2 is of both environmental and economic importance. Among the various alternative approaches to produce H2O2, the direct reaction between hydrogen and oxygen to H2O2 is conceptually the most straightforward and therefore the most attractive one. However, despite a long history and significant R&D efforts by many academic groups and industrial companies, the process still has a long way to go before reaching the commercial scale. A widespread production of H2O2 from hydrogen and oxygen in small/medium scale plants, integrated with its direct use in catalytic selective oxidation, is a good example of the future direction of sustainable chemistry [3]: on-site H2O2 production would significantly boost the use of H2O2 in all fine chemical productions, with a relevant positive impact in terms of reduction of waste, improvement of eco-compatibility, safety problems and working conditions.
In 1914, Henkel and Weber [4] were the first to propose for this process the use of noble metal catalysts. However, the low H2O2 selectivity and in parallel the high selectivity for water formation was, and still is, a major obstacle to its industrial feasibility. H2O2 formation is involved in a scheme of parallel and consecutive reactions (Figure 1) where the catalyst leading to H2O2 (reaction 1) is also responsible for its decomposition. In fact, H2O2 is unstable with respect to both hydrogenation (reaction 2) and decomposition (reaction 3), while water formation is by far the most thermodynamically favored reaction between hydrogen and oxygen (reaction 4). Therefore, the challenge has always been to find a catalyst capable of maximizing reaction (1) while depressing paths (2), (3) and (4):
As a result, this process is a typical example in which the activity and selectivity of catalysts should be precisely balanced in a framework of high efficiency [5]. This selectivity problem has not yet been solved in a satisfactory manner and still represents one of the major barriers to the use of H2O2 prepared from hydrogen and oxygen.
Furthermore, the direct contact between H2 and O2 constitutes a significant hazard [6,7] because H2/O2 mixtures are explosive in a wide range of compositions, hence operating under intrinsically safe conditions is a very important issue for the viability of the process. The flammability limits of H2 in O2 are between 4% and 94%, implying that intrinsically safe mixtures are very diluted. Clearly, operating under safe conditions results in a drop of the H2O2 productivity to industrially unacceptable values [8]. For all these reasons, the process has not yet been commercialized [5,9,10,11,12,13], despite the efforts of several chemical companies that believe the development on an industrial scale would be a real breakthrough in oxidation technologies. Typical reaction conditions are: diluted H2/O2 mixture feed gas, supported noble metal catalysts, acid promoters, methanol as solvent, near 0 °C reaction temperature, atmospheric or positive pressure and gas–liquid–solid three-phase reaction. The safety can be improved mainly by adding an inert gas, using alternative solvents such as supercritical CO2, limiting the hydrogen amount to less than 4% and using membrane catalysts.
All in all, even if the direct synthesis is in principle the simplest method to form hydrogen peroxide, it is still a “dream process” that needs a “dream catalyst”. The aim of this review is to see whether this “dream catalyst” is at hand analyzing the different proposals present in the recent literature and making evident the role of the following factors: (i) the choice of the metal; (ii) the metal promoters and alloys used to improve the activity and/or the selectivity; (iii) the role of different supports and their acidic properties; (iv) the addition of halide and other promoters to inhibit undesired side reactions; (v) the effects of particle morphology; (vi) the effects of different synthetic methods on the catalyst performance. Like in a puzzle (Figure 2), all these features should appropriately combine together to define the “dream catalyst”.
2. Choice of the Metal
Since 1914, Pd has been known to catalyze the liquid-phase oxidation of H2 by O2 to produce H2O2 and to date almost all catalysts for this reaction are still based on Pd. It has to be pointed out that the anthraquinone process, presently used for hydrogen peroxide manufacture, already includes a few percent Pd supported catalyst for the hydrogenation of ethyl-anthraquinone to the corresponding anthrahydroquinone, which implies that the direct synthesis based on a Pd catalyst will not lead to overall extra Pd consumption. The modern era of research into the catalytic direct synthesis of H2O2 started in 1967 with patents from Imperial Chemical Industries (London, England) [14] and in 1976 from Tokuyama Soda (Tokyo, Japan) [15]. In both cases, a silica supported Pd catalyst was used in an aqueous medium containing HCl and H2SO4. Later, Gosser and Schwartz [16] in 1988 reported that solutions of H2O2 in concentrations higher than 35 wt % could be produced using supported Pd catalysts at high pressure and inside the H2/O2 explosion regime.
The Pd oxidation state necessary to maximize H2O2 formation has been the object of some debate [17]. Early reports by Pospelova et al. in 1961 claimed that Pd20 clusters were responsible for H2O2 formation [18] and later other authors confirmed that Pd in a reduced state is more active and selective for H2O2 formation [19]. Conversely, Choudhary and coworkers were the first to report evidence that Pd2+ is the active center forming H2O2 because it can molecularly adsorb dioxygen yielding surface peroxo species, while Pd0 favors decomposition or over-hydrogenation of the peroxide [20].
From theoretical studies on H2O2 synthesis over metallic catalysts, Sellers and co-workers [21] suggested that the formation of H2O2 from H2/O2 mixtures is more favorable on Au and Ag than Pt and Pd. In particular, Au was identified as the potentially most selective metal among the noble metals on the basis of a comparatively high stability of hydroperoxide species on Au surfaces. A first practical example of Au catalysis for H2O2 direct synthesis was reported in a patent in 2002 [22]. Various supports were evaluated for Au nanoparticles [23]. The catalytic behavior of these systems was also studied by theoretical approaches, revealing that small Au nanoclusters (1–13 atoms) can indeed act as sites for the synthesis of H2O2 [24,25,26,27,28]. A correlation was found between the rate of H2O2 synthesis and the mean Au particle size. Okumura et al. [29] demonstrated that highly dispersed Au supported on inert materials, such as activated carbon, MCM-41 and SiO2 are effective in catalyzing the direct synthesis of H2O2, while Au supported on a basic oxide (MgO) or on a highly acidic oxide (SiO2-Al2O3) are poorly selective for H2O2 formation.
Another example of monometallic Au catalysts for the direct synthesis of H2O2 was reported by Ishihara et al. [30] who showed that a 1 wt % Au/SiO2 catalyst tested in the absence of halide promoters exhibited promising H2O2 synthesis activity (with 30% H2 selectivity) if compared to pure Pt, Pd or Ag on silica catalysts where no H2O2 was observed under identical reaction conditions. The rates of H2O2 synthesis and hydrogenation/decomposition were compared for monometallic Au catalysts with different support materials and it was concluded that basic oxides such as MgO and ZnO were unsuitable supports while Au/SiO2 performed the best.
3. Effect of Metal Promoters and Alloys
It has to be pointed out that current research in this area focuses mainly on bimetallic catalysts rather than single metal catalysts. Defective Pd sites are currently considered as the major species responsible for hydrogen peroxide decomposition, hence intense research has been done to reduce these sites by addition of a second metal mostly present as an alloy component. In particular, bimetallic systems, such as Pd-Au and Pd-Pt, have been extensively investigated. It is well known that alloying or combining two metals can lead to materials with specific chemical properties due to an interplay of “ensemble” and “electronic” effects [39], and that a bimetallic surface can exhibit catalytic properties that are very different from those of the individual metals. Enhanced productivity and selectivity were observed upon addition to Pd of another noble metal, more often Au [40,41,42,43,44,45,46]. It has also been demonstrated that the combination of Pd with Ir [47], Ag [48,49], and Pt [50] can improve both the productivity and the selectivity of the process. On the contrary, the presence of Rh and Ru increased H2O2 decomposition activity of Pd catalysts [51]. Other authors investigated Ru-Au, Ru-Pd and Ru-Au-Pd catalyst for H2O2 synthesis [52] but again results were not satisfactory.
It has been reported that the H2O2 selectivity can increase using bimetallic Pd-Ag catalysts [53] on an activated carbon support. The Ag additive increased the presence of monomeric Pd sites, which were the primary active sites for H2O2 formation, and reduce contiguous Pd sites. In addition, the content of Pd2+ was increased via the electronic interaction between Pd and Ag, which prevented the decomposition and hydrogenation of H2O2. The decreased activity was attributed to the reduced adsorption capacity of H2 and O2 due to the Ag additive. The optimal bimetallic Pd-Ag catalyst achieved a H2O2 productivity of 7022 mol kgPd−1 h−1 and a high selectivity (70.9%), which were superior to those of the Pd/C catalyst.
As to Pd-Au catalysts using mild reaction conditions, i.e., room temperature and atmospheric pressure, it has been demonstrated [41], for both plain and sulfated zirconia and ceria supports, that the addition of a 1:1 amount of gold to a monometallic Pd sample improves the productivity and especially the selectivity of the process. In these samples, gold must be in close contact to Pd, as its presence deeply changes Pd dispersion as well as its morphology and charge.
Pd-Au catalysts have been extensively studied by Hutchings and co-workers [23], making evident an increase in selectivity towards H2O2 if compared to Pd only catalysts. A ratio of Pd:Au of approximately 1:1 (w/w) provided the best performance [54]. A core-shell morphology, consisting in an Au core and a Pd-rich shell, has been identified when TiO2 [55], Fe2O3 [56] and Al2O3 [45] have been used as supports. Activated carbon was also used as a support for Pd-Au catalysts [57]. The Au present in these systems acts as an electronic promoter for the Pd rich surface of the Pd-Au nano-crystals. Acid pre-treatment of the activated carbon prior to Au and Pd impregnation resulted in a catalyst with very high selectivity (up to 95%). The high activity of this catalyst was attributed to an increase of smaller, Pd rich nanoparticles 2–6 nm in size whose number was much higher in the acid pretreated catalysts compared to a untreated one. Further studies on this material indicated that the oxidation state of the Pd in the Pd-Au alloy also plays a key role [58]. In the acid pretreated samples, the enhanced activity is associated with a higher surface concentration of palladium as Pd2+. The surface ratio of Pd0/Pd2+ was found to be an important factor in controlling the subsequent hydrogenation of H2O2. According to Goodman et al. [59], the role of Au in Pd-Au catalysts is to isolate single Pd sites that facilitate the coupling of critical surface species to H2O2, thus inhibiting the formation of by-products.
Ishihara et al. [60] prepared Pd-Au bimetallic catalysts with different Au compositions and found that the H2O2 formation rate increased with increasing concentration of Au mainly because of a decrease in the decomposition rate of H2O2. DFT calculations [61,62] indicated that the competition between the dissociation of H2O2 and the desorption of H2O2 on the catalyst surface determines the reaction selectivity. Au atoms on the surface of Pd-Au alloy can weaken the interaction of the metal surface with H2O2, thus facilitating the release of H2O2 and suppressing its decomposition.
Similar improvements in performance for bimetallic Pd-Au/catalysts were observed also with TiO2 [55] and ion exchange resins supports [63]. Even in the absence of additives, selectivities up to 80% depending on the reaction conditions were obtained.
Han and coworkers [64] suggested that, in the Pd-Au alloy catalyst, the Pd atom surrounded by Au atoms is the best active site to synthesize H2O2. Through DFT calculations, Staykov et al. [65] found that Au atoms on the Pd-Au bimetallic catalyst surface can prevent oxygen dissociation, thereby increasing the H2O2 selectivity. By DFT calculations of Pd and Pd-Au surfaces, these authors proposed the presence of surface O2 as superoxide as a favorable intermediate towards H2O2 formation. The O2 dissociation leading to H2O formation was proposed to occur mainly on Pd(111) surfaces. The presence of Au atoms could block O2 dissociation leading to higher H2O2 selectivity. A similar conclusion was drawn also by Ham et al. [66].
Han et al. [67] in a comparative study of Pd, Au and Pd-Au alloy catalysts, found that electron transfer exists between Au and Pd in the Pd-Au alloy catalyst. The role of Au in regulating the electronic structure of Pd may be one of the important reasons why Pd-Au alloy catalysts have a higher selectivity for H2O2 synthesis.
Gudarzi et al. [68] investigated the promotional effects of gold in Pd-Au bimetallic catalysts prepared by simultaneous co-impregnation on activated carbon cloth. Almost all of the Pd-Au bimetallic catalysts were more selective than Pd monometallic catalysts. The authors concluded that: (i) increasing the amount of Au resulted in more selective but less active catalysts; (ii) increasing the amount of Pd resulted in more active but less selective catalysts; and (iii) the morphology of Pd-Au particles was also affected by the amount and ratio of gold and palladium in the catalysts. These same authors concluded also that the presence of palladium oxide makes the catalysts more selective and active in H2O2 production and less active in water production if compared to the corresponding catalysts containing mainly zero-valent palladium.
In 2019, in situ X-Ray Absorption Spectroscopy was used [69] for an investigation on Pd and Pd-Au silica supported samples during the direct synthesis of hydrogen peroxide in a high-pressure gas-liquid-solid microreactor. The authors concluded that the absence of Pd-hydride in the bimetallic catalysts reduces further hydrogenation of hydrogen peroxide to water, thus increasing selectivity.
Also for Pd-Pt alloy catalysts, electron transfer takes place between Pt and Pd [50,70,71]. The introduction of a small amount of Pt into the Pd catalyst may result in electron transfer from Pd to Pt, reducing the strength of the Pd-O bond, thereby inhibiting the dissociation of O2 and increasing the selectivity of H2O2. However, when a higher amount of Pt (Pd/Pt < 8) is introduced, an enrichment of Pt on the surface was observed producing an adverse effect on the catalytic performance [70]. Adsorbed dioxygen was proposed as the precursor for the formation of -OOH surface species that react with H2 to form H2O2. According to Xu et al., excess Pt may destabilize -OOH and favor H2O2 decomposition [70]. Best results were obtained with a Pd16Pt1 sample.
Similarly, Hölderich and co-workers [72,73] found that addition of Pt to a Pd/TS-1 catalyst stabilized surface Pd2+ oxidation state. Furthermore, the metal particle shape can also be controlled by varying the Pd/Pt composition in the catalyst and/or by specific reduction conditions. An optimum level of Pt is critical for the desirable increase of surface Pd2+ concentration, but at the same time also for the undesirable changes in the surface morphology of Pd aggregates, from needle-shaped to spherical [74].
Strukul and coworkers [75] demonstrated from TEM analysis of Pd and Pd-Pt catalysts that Pd-only samples have well-developed spherical particles, whereas Pt-Pd bimetallic samples contain irregularly shaped particles. The catalyst with the smallest particles and a spread in the size distribution was found to be the most active/selective in the direct H2O2 synthesis. These studies demonstrate that the effect of the addition of Pt to Pd to increase the yield of hydrogen peroxide is sensitive to the Pt amount: only using a low Pt content, it is possible to improve H2O2 selectivity (from 55% to 70%) and productivity with respect to the monometallic sample [76].
Headwater Technology Innovation developed a technology for the synthesis of H2O2 directly from H2 and O2 [77] based on catalytic system called NxCatTM, where the active components are Pd-Pt particles with a uniform 4-nm size feature. This enables a high production of H2O2 with a 4% hydrogen in air (i.e., outside the flammability range) and also maximizes the H2O2 selectivity up to 100%.
Hutchings et al. [78,79] were the first to study Pt-Au-Pd trimetallic catalysts and found that introducing a small amount of Pt into a Pd-Au bimetallic alloy can suppress the hydrogenation and decomposition of H2O2, increasing the yield of H2O2.
Metals other than Au and Pt were also tested as promoters to improve the performance of Pd catalysts in the direct synthesis of H2O2. In 2016, a bimetallic sample containing a non-noble metal (Ni) was also proposed [80]. The Ni0.1Pd0.9 catalyst was reported to show three times higher activity compared to the sample with Pd alone and proved stable for 72 h. More recently, on the basis of density functional theory (DFT) studies, Xu et al. reported the possible increase of H2O2 production by using different metals such as tungsten, lead and molybdenum [81]. In 2018, Huang et al. have proposed RuNi (111) samples as good candidates for hydrogen peroxide direct synthesis due to their self-activation mechanism [82].
In addition, the addition of zinc was found to improve the catalytic performance of palladium catalysts supported on alumina [83] and, more recently, in a Zn-CNTs-O2 system [84] and on silica [85]. The promotional effect of Zn was attributed to both particle geometric changes and to an electronic interaction between Pd and Zn. The addition of Zn increases the isolated Pd sites, recognized as the primary sites for H2O2 formation, while reducing the contiguous Pd sites, effectively suppressing the H2O formation from H2 and O2. On the other hand, the electronic effect of zinc leads to a higher Pd dispersion and Pd0 content than in monometallic Pd, thus increasing H2O2 hydrogenation. With the optimum bimetallic catalyst (Pd/Zn 1/5), a better H2O2 productivity than with the monometallic catalyst was found (25,431 vs. 8533 mol kgPd−1 h−1).
Almost 100% selectivity toward H2O2 was reported for a Pd-Te catalyst supported on titania with the Pd/Te atomic ratio of 100:1 [86]. By DFT calculations, the role of Te in restraining side reactions was suggested. The formation of a bimetallic Pd-Te surface is believed to be the origin of the active sites for the catalytic production of H2O2. By suppressing the dissociative activation of O2 on Pd, Te plays a critical role in enhancing the catalytic performance for H2O2 synthesis. On the other hand, the addition of Te has also a promoting effect on the dispersion of Pd particles on TiO2, leading to an increase in low-coordinated active sites at corners and edges by reducing the particle size; these sites are much more active for the undesired production of water. Thus, the Pd/Te ratio should be finely tuned to balance the ratio of terrace Pd-Te sites and the particle size for the best performance.
With the aim of stopping H2O2 hydrogenation, a tin-based Pd alloyed catalyst was developed. The latter exhibited a selectivity higher than 95% [87].
Very recently [88], the addition of a second non precious metal (Sb) was evaluated. Antimony promoted the dispersion of Pd on TiO2. The increase in Sb concentration led to an increase of isolated Pd sites while the amount of contiguous Pd sites that are highly reactive for H2O2 hydrogenation were reduced. As a result, the Sb modified Pd surfaces significantly enhanced the non-dissociative activation of O2 and therefore H2O2 selectivity. A value of 73% of selectivity was claimed working at atmospheric pressure.
The higher activity of a Pd-Hg bimetallic catalyst with respect to a Pd sample was suggested by DFT calculations [89]. The presence of Hg on Pd(111) results in geometric and electronic synergisms with respect to O2 absorption. DFT calculations, employed to investigate O2 adsorption on the Pd and Pd-Hg alloy surfaces, suggested O2 adsorption occurring via either a superoxo or a peroxo pathway and, when Hg is alloyed to Pd, there are more surface adsorbed superoxo groups compared to adsorption on a monometallic Pd surface.
As is clear from the above discussion, there is no uniform trend as far as Pd promotion is concerned. We have tried to summarize the behavior of the most interesting metal promoted/alloyed catalysts in Table 1.
We have homogenized the parameters reported by the different authors using the same units even if in the original paper they are different. Even so, trying to establish a ranking with respect to activity, selectivity, productivity among these catalysts is almost impossible because of the extreme variety of reaction conditions, catalyst composition, etc. Moreover, the effect of the metal promoter in the individual cases is almost always mixed with those of other factors such as the different supports, the acidity of the medium and/or the support, the presence of halide promoters, etc., which will be dealt with in the next sections. As is clear from Table 1, in many cases, even high average activities can end up with moderate to poor overall productivities because of a rapid catalyst deactivation.
A picture of the current trend with respect to the metal promoters used to improve the performance of Pd catalysts is schematically represented in Figure 3. What we can say to summarize this section is that, among the wide range of metal promoters tested to improve the performance of Pd catalysts, prominent results have been especially observed with Au.
A general suggestion that emerges from these studies and is common to most metals tested (Figure 4) is that the role of the second metal is mainly to improve Pd site isolation and increase its oxidation state. Both effects will favor molecular adsorption of dioxygen increasing the surface concentration of surface peroxo species as precursors for H2O2 formation, while at the same time depressing dissociative O2 chemisorption and hydrogen peroxide O–O bond breaking, two conditions that lead to direct water formation and H2O2 decomposition, respectively.
4. Effect of Supports
As is intuitive, an important role is played by the support with its ability to modulate the electron density on the metal and its capacity to impart the appropriate metal morphology. Many different supports have been investigated for this reaction, even if the most common ones are carbon [57,90,91] and silica [92]. A typical feature of carbon is its essentially hydrophobic nature. This may have a protective role towards hydrogen peroxide, as it is known that this reagent is more stable to decomposition in organic solvents rather than in water. Furthermore, the support can stabilize the presence of oxidized active sites and this in turn might have an important influence in decreasing H2O2 decomposition reaction.
Besides carbon, simple oxides or mixed oxide supports have been tested such as, for example, ZrO2, CeO2, ZrO2-CeO2, TiO2, Al2O3, Ga2O3, SiO2, either plain or modified with promoters aiming to change their acidic properties to disfavor H2O2 decomposition. Since the palladium oxidation state represents an important issue in H2O2 decomposition, a support with redox properties, such as CeO2, can actually contribute to the stabilization of oxidized Pd sites [41]. Many of these supports have been synthesized employing precipitation and sol-gel techniques.
A comparison among monometallic palladium based samples over different supports have shown that, in a semi batch reactor under mild conditions, Pd/SiO2 gives the highest selectivity and productivity [93], while catalysts supported on sulfated zirconia and ceria performed better by continuous operation in a trickle bed reactor [94].
As to bimetallic catalysts, Edwards et al. found that a carbon supported Pd-Au catalysts give the highest reactivity, while Pd-Au samples on TiO2 and SiO2 performed better than that on Al2O3 and Fe2O3 [95] under the same experimental conditions. It was proposed that the isoelectric point (IEP) of the support has a major role in improving the selectivity towards the formation of surface peroxide, by controlling the surface charge of the catalyst.
Various nanostructured carbons (activated carbon, carbon nanotube, carbon black, and ordered mesoporous carbons) were investigated as support for Pd-Au samples [96]. The results demonstrated that a large fraction of oxygen functional groups on the carbon surface was an important issue in the synthesis of highly dispersed bimetallic catalysts with actual Pd-Au alloying, this being a pre-condition for high H2O2 selectivity. Microporous carbons like e.g., activated carbon suffered from mass transfer limitations, leading to significantly lower H2 conversion than mesoporous catalysts. According to Yook et al., hydrogen peroxide produced from a Pd-Au catalyst in the micropores had a higher tendency to subsequent disproportionation/hydrogenation into water due to retarded back-diffusion of hydrogen peroxide leading to decreased selectivity. These authors concluded that only mesoporous carbons with a high oxygen content are highly desired as supports for the Pd-Au bimetallic catalyst, a condition necessary to achieve high catalytic activity and selectivity.
A series of Pd catalysts supported on ordered mesoporous carbons [97] were prepared by surfactant templating and incipient wetness impregnation of ordered mesoporous carbons that were previously synthesized at different temperatures (T = 8, 15, 25, 35, and 45 °C). Conversion of hydrogen and hydrogen peroxide yield showed volcano-shaped behaviors as a function of the preparation temperature when halide and acid additives were present (Figure 5). The catalytic performance was found to be closely related to the palladium surface area. In an attempt to avoid acid additives, a series of palladium catalysts supported on heteropolyacids embedded in an ordered mesoporous carbon were prepared by incipient wetness impregnation and ion exchange methods and changing the heteropolyacid content (5, 10, 20, 30, and 40 wt %). Selectivity and yield for hydrogen peroxide showed again volcano-shaped behaviors with respect to the heteropolyacid content. It was observed that the hydrogen peroxide yield increased with increasing acidity of the catalyst. The latter, along with the capacity to control the surface area of the carbon support, was a key factor to improve the catalytic performance.
Ordered mesoporous carbons with a CMK-3 structure probed to be better supports for Pd-Au nanoparticles compared to non-ordered mesoporous carbon [98]. The pretreatment of both the supports with acid enhances the metal dispersion (2–3 nm) and promotes the gold enrichment of the smaller nanoparticles, decreasing H2O2 decomposition. The optimal composition corresponds to an Au/Pd ratio in wt % of 1. Selectivities up to 99% can be obtained.
TiO2 was found a good support for Pd nanoclusters giving high selectivities towards H2O2 [99]. The possibility to use TiO2 nanowires as supports was also investigated. Indeed, the performance of a support is strongly related not only to its crystalline phase and the extent of crystallinity but also to the surface structure of the material. In a number of practical applications, it has been shown that the performance of one-dimensional nanostructures (such as nanofiber-based materials) is competitive with (and in some cases outperforms) their conventional 0-dimensional counterparts (such as nanoparticle-based materials) [100]. However, their very high activity results mainly in the formation of water so that catalysts supported on TiO2 nanoparticles turned out to give higher selectivity and higher TOF than the materials based on TiO2 nanowires.
Strukul et al. [101] studied the effect of the textural properties (pore size distribution and wall thickness) of mesoporous silica supports such as MCM-41 and SBA-15 on the activity and selectivity of Pd-based catalysts, observing a correlation between the textural properties of the support and the catalytic activity of the examined samples. In the case of MCM-41, the samples show high activity but poor selectivity as well as poor stability. Because of the narrow pores (2.9 nm), only Pd nanoparticles of about 2 nm or less were observed inside the channels while a fraction of large Pd particles (large in weight but small in number) was found outside. The small Pd nanoparticles were very active due to the presence of highly energetic Pd sites (defects, edges and corners), but, somehow expectedly, caused a drop in selectivity because these energetic sites trigger H2O generation through dissociative O2 chemisorption. In addition, the thin wall thickness of the MCM-41 makes it poorly stable. The problems were solved with SBA-15 where the catalysts performed very well in terms of activity and selectivity. The relatively large pore size of SBA-15 is beneficial to obtain a catalyst with a suitably larger Pd average particle diameter (4.5 nm), a likely good compromise between a high dispersion and the presence of less energetic sites leading to high activity avoiding a drop in selectivity. Almost 90% of the palladium present was usefully located inside the pores. The relatively large pore size of the SBA-15 also allows an easy diffusion of reagents and products while the thicker wall thickness of the SBA-15 imparts a good mechanical stability and reusability.
In 2018, a series of Pd-Au catalysts supported on a mesoporous functionalized silica (SBA-15) was investigated [102]. Support functionalization was performed using different groups (SO3H, NH2 and SH) while the metal was introduced by ion exchange. Compared to a Pd-Au system over non-functionalized SBA-15, catalytic performance is improved upon functionalization, increasing hydrogen peroxide rate in sulfonated SBA-15 systems and reducing hydrogenation/decomposition activity by adding amine groups. Organic functional groups with acid-base properties acted as anchoring sites controlling both the dispersion of the metallic active phase as well as the oxidation state of gold and palladium species, present as Au+ and Pd2+. The presence of amine groups suppresses the support microporosity and probably influences the metal cluster size. Functionalization of a mesoporous silica SBA-15 support by means of mercapto/sulfonic groups allows controlling the Pd-Au dispersion and therefore the particle size distribution. The presence of oxidized Pd and Au centers are particularly important to depress the hydrogenation/decomposition activity. Again, the average metal particle size achieved in these samples was a good compromise between a high metal dispersion necessary for high catalytic activity and the presence of less energetic sites, on which O2 can chemisorb without dissociation.
Pd (0.3 or 0.6 wt %) was supported also on zirconia promoted with rare earth oxides (Y2O3 and La2O3) [103]. These materials were chosen because of their capability to increase both the metal dispersion and the O2 mobility (which favors the active metal activity). Larger Pd particles were obtained upon supports with smaller pore diameters. The larger Pd particles were more selective towards hydrogen peroxide, again confirming that highly unsaturated sites, such as Pd atoms at corners or edges largely present on small particles, cleave the O–O bond, leading to water. The authors concluded that highly reducible supports giving larger metal particles are most desirable in the preparation of catalysts for the H2O2 direct synthesis.
In Table 2, we report a comparison among different Pd and Pd/Au catalysts dispersed on different supports. Among the large number of possible catalysts and supports reviewed above, we have deliberately chosen those tested under relatively similar conditions. As is clear from the table, even in this limited set of data, no support per se seems to impart special properties to the catalyst or seems to outperform the others in terms of promotion of activity and selectivity. Key features for supports seem to be the ability to increase Pd oxidation state and, to some extent, particle size, two factors that for the reasons exposed above improve the catalyst performance.
5. Effect of the Acidity
From the above discussion, it already appears that the acidity of the support, either intrinsic or induced by post treatment, is a key issue and has been the subject of intensive research. Since the early days of research in this area [16], the use of media containing acids and/or halide promoters has been a common practice both to stabilize the hydrogen peroxide produced and to improve the catalyst performance. This, however, implies some corrosion problems, especially in view of possible applications on a large scale, and the use of these additives should be avoided as much as possible. This is why a large variety of acidic supports have been tested in H2O2 synthesis, especially in recent years [104,105,106,107,108,109,110].
Park and Fierro et al. [99,111,112,113,114,115] studied the performance of Pd catalysts supported on silica functionalized with sulfonic groups and found that the SO3H functional groups not only can promote the generation of H2O2 but also make it possible for the process to be operated using less inorganic acid, thus reducing reactor corrosion and the loss of active metal. The role of sulfonic groups was tested also in the case of sulfonated cation exchange polystyrene resins. Catalysts prepared by anchoring Pd2+ ions onto these organic supports were particularly effective with methanol as a solvent [114]. Lee and coworkers [115] operated the direct synthesis of hydrogen peroxide in continuous by using SO3H functional resin as catalyst support, and the concentration of the H2O2 solution reached 9.9 wt %. The high performance of these catalytic systems results from the ability of the sulfonic acid groups of the resin to interact with and stabilize the Pd2+ ions without further reduction to metallic palladium [116].
Edwards et al. [11,117] systematically reviewed the use of acidic supports and pointed out that the isoelectric point of the support was a key factor in the stabilization of H2O2 under reaction conditions. Supports with low isoelectric points such as carbon materials are beneficial for the stabilization of H2O2. In particular, supports that have lower isoelectric points, such as carbon, provide much lower rates of H2O2 degradation than those catalysts utilizing a support of higher isoelectric points such as magnesium oxide. The same group found that their best catalyst was a bimetallic Pd-Au deposited on HNO3 or HOAc washed carbon [49,54,56,118]. The second best support being CeO2, even if in this case the monometallic catalyst was more productive than the bimetallic one but to the detriment of selectivity that was much lower (43%).
Similarly, Pd-Au catalysts prepared by incipient wetness co-impregnation on different supports have been compared [119]. The productivity follows the trend: Pd-Au/SiO2 ≈ Pd-Au/sulfated ZrO2 > Pd-Au/ZrO2 >> Pd-Au/CeO2 ≈ Pd-Au/sulfated CeO2. The presence of Pd-Au alloy particles has been observed on all samples and their characterization indicate that there is an effect of the acidity of the support on both morphology and size of the bimetallic phase. More specifically, the more acidic was the support, the larger was the size. However, HRTEM and DR UV-Vis spectroscopic analysis showed that only Pd-Au/SiO2, Pd-Au/sulfated ZrO2 and Pd-Au/ZrO2 contain bimetallic particles exposing the active less energetic sites. In contrast, the small bimetallic nanoparticles observed on ceria expose only more energetic sites responsible for the formation of water.
Among solid acid supports, regularly cited examples include acidic supports such as MoO3 on zirconia, V2O5 on zirconia, supported sulfuric acid catalysts [120,121] and fluorinated alumina [20].
It is known that mixed metal oxides such as SiO2-Al2O3 and SiO2-ZrO2 can have higher surface area and larger surface acidity than pure metal oxides. Park and co-workers [110] investigated the use of TiO2-ZrO2 as a support in the synthesis of H2O2. A number of catalysts were produced varying the ZrO2 content. A volcano shaped trend with respect to the ZrO2 content was observed, with the catalyst having 75 mol% ZrO2 showing the largest surface area as well as the largest surface acidity and the highest H2O2 yield (ca. 6.5%).
Some authors [122] investigated doped mesoporous SBA-15 catalysts demonstrating that the use of Al as dopant leads to an enhancement in catalytic productivity and selectivity. This was attributed to an increase in the number of Brønsted acid sites in the support framework.
Park and co-workers [104] compared a series of Pd/H-ZSM-5 supported catalysts with varying Si/Al ratio to investigate the effect of the Brønsted/Lewis acid sites ratio. By increasing this ratio, the yield of H2O2 also increases with an effect equivalent to adding a strong acid additive to the reaction solution. Indeed, the catalyst exhibiting the largest Brønsted acidity (30/1 Brønsted/Lewis acid sites ratio) provided also the highest H2O2 yield.
Sun et al. [123] demonstrated that the Keggin-type heteropolyacid tungstophosphoric acid can be utilised as a support for Pd to produce an active catalyst without the need of acidic promoters. It was shown that this Pd only catalyst showed larger selectivity towards H2O2 compared to more conventional Pd only catalysts.
Similar results were observed by Park et al. in Pd exchanged heteropolyacid catalysts using various alkaline metal ions [109]. It was found that the catalyst incorporating Cs showed the highest rates of conversion and overall H2O2 yield with respect to those incorporating K and Rb. Further studies by the same group on a series of Pd/heteropolyacid with varying Cs content [124] showed that the most acidic catalyst tested was also the best performing one. Similar results were observed also when Cs-exchanged heteropolyacids were embedded on a Pd supported on a mesostructured cellular foam silica catalyst finding a correlation between yield of H2O2 and acidity of the catalyst. Hutchings and co-workers have investigated Pd–Au catalysts both exchanged with and supported on Cs-containing heteropolyacids [107] and evaluated these catalysts for the direct synthesis of H2O2 over a range of reaction conditions. When compared to the previously reported highly effective Pd-Au catalyst on activated carbon or metal oxide supports under conditions that are more favorable for the environment and the process economics, both the exchanged and supported Pd-Au heteropolyacid catalysts resulted in being much more active.
The use of acidic supports such as e.g., zirconium oxide [125] and niobium oxide [126] for the preparation of catalysts used in the direct synthesis of H2O2 has been the subject of some industrial patents where improved selectivity and activity were reported.
The issue concerning the hydrophilic/hydrophobic properties of the support was also addressed in some papers. Fu et al. [127] reported that Pd deposited on hydrophilic supports is very efficient for H2O2 decomposition. However, H2O2 decomposition was slowed down when Pd was deposited on a hydrophobic support and then mixed with a small amount of Pd free hydrophilic component. Several patents [128,129] also indicated that Pd catalysts supported on materials having an intermediate philicity character (e.g., fluorinated carbon) are suitable for the direct synthesis of H2O2 because of their low activity for H2O2 decomposition.
The examples reported above and Table 3 demonstrate beyond any doubt the beneficial effect of an acid environment on the selectivity towards hydrogen peroxide, this being associated with a stabilization of the newly formed H2O2 on the catalyst surface (Figure 6). On this view, there is a general agreement as it is quite well known that hydrogen peroxide is more stable at low pH (commercial H2O2 solutions are stabilized with sulfuric acid). However, the role of acidity could be more complex and include also some complementary explanations. For example, an influence of the acid support material on the electronic structure of the metal particle, as suggested by several authors [130,131,132,133]. Acidic supports normally increase the electron deficiency on the noble metal [130,134] and this modified electronic structure may influence the chemisorption of reactants and consequently the catalytic properties of the metal particles. Moreover, Sachtler and coworkers, studying a series of Pd catalysts supported on different zeolites, suggested the formation of metal-proton adducts on Brønsted acidic sites [135,136], very active in protonating hydrocarbons such as neopentane. These Pd-proton adducts may have also an active role in hydrogen peroxide synthesis, e.g., protonating surface peroxides as was suggested by Abate et al. some years ago [137].
6. Effect of Halide Promoters
The use of halide anions (F−,Cl−,Br−,I−) as promoters has long been recognized to improve the activity and/or selectivity in the direct synthesis of hydrogen peroxide. Their incorporation has been achieved either by impregnating the halide ions into the supported Pd catalysts [138,139,140,141] or via the deposition of halide ions onto the support prior to the deposition of Pd [31,32,33,140,142].
Already in 1993, Hiramatsu et al. [120] disclosed that halogen ions could be adsorbed into supported Pd catalysts by impregnation with a halogen source. Choudhary et al. [143] claimed that incorporating two halogens in a Pd catalyst can create a strong synergistic effect, thus increasing H2O2 productivity. The first halogen is generally bromine while the second halogen can be either fluorine or chlorine.
Strukul et al. [33] investigated the catalytic performance of Pd catalysts supported on SO42−, Cl−, F−, and Br− doped zirconia in H2O2 synthesis under mild conditions (20 °C, 1 bar). It was found that a doped support could generally improve the production rate and the selectivity towards H2O2, especially in methanol as solvent. Best overall H2O2 selectivities were observed with F− and Br− dopants, followed by SO42−, then by Cl− and the non-doped sample. By using kinetic analysis, it was observed that the rate constants for the undesired H2O2 hydrogenation were about the same for the SO42−, F−, and Br− doped samples as well as for the non-doped sample, meaning that the undesired hydrogenation of H2O2 is affected more by the acidity of the environment and to a lesser extent by the doping anions, in agreement with the observation of Lunsford et al. [32]. Hence, the selectivity difference among the various samples is determined essentially by the competition between H2O2 and H2O direct formations. Cl− doping leads to the highest H2O2 formation constant, but, at the same time, the maximum rate towards H2O generation, thus resulting in bad selectivity.
Lee and coworkers compared the results of hydrogen peroxide synthesis using Pd nanoctahedrons of varying sizes with bromide additives [144]. Regardless of the presence of bromide additives, hydrogen peroxide selectivity was found to increase. The bromide ions were selectively adsorbed at the water-forming energetic sites, thereby enhancing hydrogen peroxide selectivity. This effect was verified by demonstrating that, as the ratio of energetic/less energetic sites increased, the number of bromide ions being adsorbed increased, thus outweighing the effect of the nanoparticle size. In addition to the steric shielding effect of the energetic sites, according to Choudhary et al., the adsorbed halide species also alter the electronic state of the Pd atoms [145,146]. These authors proposed a reaction mechanism where electron charges donated from the adsorbed bromide ion is transferred through the Pd to the adsorbed oxygen molecule, which in turn reacts with a proton to form a hydrogen peroxide molecule [147]. Generally, halide ions are added in the form of alkali salts (e.g., KCl, KBr, NaCl, and NaBr) or acid (HCl, HBr, etc.). The cations (K+, Na+, Cs+, etc.) from the alkali salts have no effect on hydrogen peroxide synthesis [148].
7. Effect of Other Promoters
Abate et al. [149,150,151] studied Pd and Pd-Au catalysts supported on carbon nanotubes finding that N doped carbon nanotube supports favor the dispersion and stability of Pd nanoparticles, thus improving the H2O2 formation rate. In addition, these authors postulated that the introduction of N functional groups would also increase the acidity of the catalyst surface, thereby leading to a stabilization of H2O2.
The surface electrostatic properties of TiO2 can be largely modulated by nitrogen doping. Miyauchi et al. [152] observed a negative shift of the zeta potential and a decreased isoelectric point (IEP) in nitrogen-doped TiO2. This result was confirmed by Li et al. [153], claiming a decreased IEP of N-TiO2 with respect to commercial TiO2. This observation suggest that nitrogen-doped TiO2 could be a better support for Pd and Pd-Au based catalysts, favoring the H2O2 direct synthesis by virtue of its lower IEP. Moreover, it is known that acid additives inhibit the formation of water, but also cause reactor corrosion and leaching of active metal from the catalyst. Still, the presence of atomic nitrogen within the support framework enhances the surface acidity of the catalyst, so that the use of inorganic acids in solution can be minimized. Taking into consideration all the mentioned aspects, N-doped TiO2 is a promising support to develop Pd based catalysts active in the direct synthesis of hydrogen peroxide.
N-doped materials gave rise to a higher selectivity than the parent undoped materials for Pd samples supported on both TiO2 nanoparticles vs. TiO2 nanowires as support. This is apparently due to the synthetic procedure, leading to larger Pd nanoparticles that are unable to dissociate the O2 molecule [154].
Hutchings et al. [155], by using an insoluble heteropolyacid support in water as the solvent, studied the effect of some metal ions (Cs+, Rb+, K+ and Ag+) finding that Cs+ and Rb+ containing catalysts were extremely active for H2O2 synthesis.
Gudarzi et al. [156] studied the factors affecting the catalytic decomposition of H2O2 and found that carbon supports containing carboxylic functional groups can stabilize H2O2 in solution and reduce its decomposition and hydrogenation. Nevertheless, carbon materials have another problem: excess surface COOH functional groups are unfavorable to H2O2 synthesis because they increase the hydrophilicity of the support, thus triggering the decomposition of H2O2 on the surface of the catalyst.
8. Influence of Surface Morphology
The effects of nanoscale modifications of Pd catalysts on the direct synthesis of hydrogen peroxide have been recently reviewed [81]. Application of nanotechnology techniques to the development of an effective catalyst can operate in two directions: (i) on the morphology and size control of the metal nanoparticles to enhance selectivity towards hydrogen peroxide formation and (ii) on the immobilization of the as synthesized nanocatalyst to minimize active metal loss during the reaction.
Indeed, it is not the support surface area but rather the morphology and surface composition of active components that play a key role in controlling the activity/selectivity of a catalyst system. This was shown unequivocally for example by comparing a Pd-Au catalyst supported on a 40 m2/g TiO2 to a 2.0 m2/g rutile-TiO2 supported one where the former was found to be less active for H2O2 formation than the latter.
In some studies, colloidal Pd was found to be mainly responsible for H2O2 formation either using a Pd/silica catalyst or PdCl2 as a salt in an acidic (HCl) solution but carrying out the reaction at a relatively high hydrogen to oxygen ratio of 1:2 [157]. In the latter case, the Pd colloid forms under reducing conditions in the acidic solution. Evidence for claiming the role of colloidal Pd came from comparing the formation rates of H2O2 in the presence of the supported catalyst and after its removal from the reaction solution. TEM provided evidence for the presence of colloidal Pd in solution, both when using Pd/silica and PdCl2. Similarly, size-controlled nanocolloidal Pd-Au (particle size between 2 and 8 nm) was reported to be a highly active and selective catalyst for H2O2 formation in an acidic solution containing NaCl and H2SO4 [60]. A higher activity was observed than with Pd-Au supported on rutile-type TiO2.
A core−shell structured Pd-SiO2 catalyst was synthesized by encapsulating Pd nanocrystals grafted on silica nanobeads with a porous silica shell [158]. The production rate of H2O2 significantly increased when using a mesoporous shell catalyst rather than a microporous one of similar shell thickness. A thinner thickness resulted in being more favorable in terms of pore-diffusion rate. However, the shell thickness must be adjusted because a very thin shell layer cannot protect the core Pd crystals from sintering. Under ambient reaction conditions, a hydrogen peroxide production rate of 1120 mmol H2O2/gPd·h was observed.
In 2017, by combining density functional theory (DFT) calculations and Sabatier analysis, a strategy was proposed to design more efficient Pd-based nanocatalysts [83]. The average number of valence electron of Pd-shell atoms (actually tuned by dopants) was identified as the intrinsic factor for controlling the activity and selectivity of the nanocatalysts. Dopants with suitable electronegativity can adjust the valence electrons of Pd-shell atoms to an optimal range to simultaneously enhance the activity and selectivity of the nanocluster. With this strategy, Pd-W, Pd-Pb, Au-Pd-W, Au-Pd-Pb, Au-Pd-Mo, and Au-Pd-Ru were predicted as potentially good candidates.
To achieve high activity and selectivity for a long reaction time, not only the active and selective state of the catalyst but also its dynamic transformations under the reaction conditions need to be identified. Accordingly, catalyst formulations with “sacrificial supports” that during reaction enable the regeneration of the active and selective state have a high potential for improving catalyst lifetime and reducing the metal loading [159]. Accordingly, the development of Pd on carbon nitride catalyst was investigated. With respect to N-containing carbon nanotubes, carbon nitrides provide a higher concentration of N sites, a flexible network of π-conjugated polymeric subunits with sp3 linking subunits, and a flake like morphology with high exposure of reactive C edge terminations. This results in a more effective kinetic stabilization of the electronically modified Pd species. Strong chemical interaction of Pd with N species and surface protection by carbonaceous impurities are the factors explaining the stable performance of these materials. Structurally optimized carbon nitrides enable lowering of Pd loading and increasing catalyst lifetime.
A further issue arises from the low H2 and O2 concentrations required to be outside the explosion limits and operate safely. Under these reaction conditions, the achievement of acceptable reaction rates and the minimization of the side oxidation reaction to water require the use of high pressures. Reactors necessary to operate on a medium-large scale, however, remain intrinsically unsafe, an aspect that represents a critical unsolved question. Recently, the use of specially designed membranes has attracted considerable interest because it provides a promising approach to overcoming, at least in principle, the problems of safety and selectivity [75]. The reason is that, by using membrane materials as the support to prepare membrane catalysts, H2O2 can be synthesized without direct contact between H2 and O2. In this respect, already ten years ago Strukul and coworkers prepared supported Pd, Pd-Pt and Pd-Ag membrane catalysts using porous alumina film, porous alumina tube or porous ceramic tube as the support [49,137]. In principle, H2 dissociates over metal active sites into hydrogen atoms that can penetrate the membrane catalyst and react with O2 on the other side of the membrane to form H2O2. At present, the main problem of membrane catalysts is their low reaction efficiency caused by the low rate of mass transfer. The performance of membrane catalysts in H2O2 synthesis can be improved by surface decoration and modification [160,161,162,163,164,165,166].
As to the Pd nanoparticle morphology, hydrogen peroxide synthesis at different Pd facets has been investigated using DFT simulations [121,167]. In fact, different crystal surfaces differ in the interatomic distances between Pd atoms, yielding different interaction for the adsorption/desorption of species such as oxygen and hydrogen. However, there is still no agreement on which facet is more favorable for hydrogen peroxide formation. Tian et al. reported that the Pd {111} facet promotes hydrogen peroxide formation, whereas the {100} and {110} facets promote water formation [168]. These researchers explained that, at Pd {100} and {110} facets, there is a bond weakening that causes the O–O bond to stretch from 0.124 to 0.413 nm. At the {111} facet, the magnitude of the bond weakening is significantly smaller and the O–O bond stretches only to 0.137 nm, accounting for the non-dissociative adsorption of O2 and a higher selectivity toward hydrogen peroxide.
Also by DFT calculations, Deguchi et al. [47] found that the adsorption energies of H2, O2, H2O2, and HBr were larger on coordinatively more unsaturated sites (such as corners and edges) than on more saturated sites such as a {111} facet. The energy profiles of the H2 + O2 reaction suggested that H2O2 would be smoothly produced on more saturated sites such as the {111} facet. On the contrary, the formation of H2O and the decomposition of H2O2 would be preferred on more unsaturated sites such as corners and edges. The opposite conclusion was drawn by Zhou et al. who reported that the {110} facet is favorable for hydrogen peroxide formation while the {100} and {111} facets are favorable for water formation [121].
Based on experimental evidence, Lee et al. demonstrated a shape-dependent catalytic activity of palladium nanoparticles for the direct synthesis of hydrogen peroxide [169]. They investigated silica-supported nanocubes and nanoctahedrons prepared by a colloidal method. Pd octahedrons were synthesized via seed-mediated growth of the corersponding nanocubes, where the morphology could be altered to that of a cuboctahedron, truncated octahedron, and octahedron, in series. The cubes and octahedrons were enclosed by {100} and {111} facets, respectively. In activity tests, the Pd octahedron presented higher hydrogen peroxide selectivity and productivity than the Pd cube. It was therefore suggested that the Pd {111} facet is more active than the Pd {100} facet in direct hydrogen peroxide synthesis.
A schematization of the most structure representative images of the catalysts is reported in Figure 7.
As far as the Pd particle size is concerned, a decrease in size and thus an increase in the specific Pd surface area are positively correlated with reactant conversion and product yield. This, however, decreases the selectivity towards hydrogen peroxide [93]. Pd surface active sites are classified either as energetic (on corners, edges, or defects) or less energetic (on terraces) depending on the degree of unsaturation. Highly unsaturated energetic sites containing a high number of available valence orbitals imply a preference for dissociative dioxygen chemisorption, leading to water formation and low hydrogen peroxide selectivity. Conversely, less energetic sites with a lower degree of unsaturation would exhibit selective catalytic activity towards hydrogen peroxide formation.
This point was clearly demonstrated by Lee et al. on their Pd nanocubes and nanoctahedrons of varying size, as they observed [144] that a larger Pd nanoparticle size, despite the decrease in specific surface area leading to lower hydrogen conversion, having fewer energetic sites, offers higher hydrogen peroxide selectivity. These authors suggested that selective blocking of energetic sites on Pd nanocatalysts can increase hydrogen peroxide selectivity.
The stability of Pd nanoparticles is another important issue that must be addressed in order to realize sustainable operations. Loss of metal after hydrogen peroxide synthesis, ranging from 25 to 77 wt %, was reported by Lee and coworkers [169,170,171]. Pd nanoparticles are removed from the support structure by physical detachment and/or chemical leaching. Choudhary and Samanta reported significant Pd chemical leaching from the catalyst when using phosphoric acid as promoter in concentration over 0.3 M. Chemically induced leaching was spectroscopically confirmed by observing a loss of uniform morphology of Pd nanoparticles [147]. On the other hand, physical detachment of nanoparticles from silica with no evidence of chemical leaching was confirmed by Kim et al. through TEM and XRD analyses when using phosphoric acid in concentration below 0.3 M [115,169]. In order to improve retention of the Pd nanoparticle, the core@shell structure is being actively studied [172,173,174]. In this structure, Pd nanoparticles are enclosed by an oxide shell during catalyst preparation, and, in the subsequent calcination, the surfactant (or polymer) used for this purpose is incinerated, creating pores while at the same time preserving the core metal size even at high calcination temperatures (Figure 8). This peculiar structure physically encloses the Pd nanoparticles to help prevent physical detachment, thus enhancing stability during reaction. Several oxides have been used as shell, namely SiO2, TiO2, Al2O3, ZnO, SnO2, Cu2O, Fe2O3, Fe3O4 and CeO2 [175,176,177,178,179,180,181,182,183,184,185,186,187]. SiO2 is a popular choice because of the relative ease of synthesis while its low isoelectric point promotes hydrogen peroxide production rate [185]. Lee et al. demonstrated superior catalytic performance of nanoparticles in the core@shell structure (Pd@SiO2, 554 mmol H2O2/gPd h) over that observed on a conventional support (Pd/SiO2, 267 mmol H2O2/gPd h) [183]. Additionally, the Pd loss was reduced to only 5%.
From the above discussion on the effects of promoters and surface morphology, the overall emerging framework indicates manifold, often intermingled, roles that contribute to define the catalyst activity and selectivity. In trying to summarize those aspects on which there seems to be a general consensus, we can name the following (Figure 9): (i) the active metal should be present in medium-sized particles (5–15 nm) as a good compromise between activity and selectivity and to avoid the presence of highly coordinatively unsaturated (defective) metal sites; (ii) promoters generally acts as poison for the defective sites, thereby increasing hydrogen peroxide selectivity; (iii) they may increase the acidity of the support stabilizing hydrogen peroxide and (iv) they can act as donors anchoring the metal to the support, thus preventing sintering, leaching and/or physical detachment.
9. Effect of Synthetic Methods
Given the importance of the active phase morphology on the outcome of the reaction, the preparation method with which the metal(s) are introduced into the support becomes a critical issue in defining the activity and selectivity properties of the catalyst, hence impacting differently the formation of hydrogen peroxide vs. its decomposition. Different authors generally try to achieve catalysts with differently sized active phase particles in order to study the influence of Pd particle dimensions both on the synthesis and on the decomposition of hydrogen peroxide. Moreover, the methodology adopted will try to reduce the defective Pd sites that seem to be responsible for hydrogen peroxide decomposition trying to conjugate high activity with high selectivity. As will be clear in the afterwards, many authors have found significant differences depending on the synthetic methodology chosen, but to date a general trend to be recommended does not seem to emerge.
In general, a homogeneous distribution of the active phase on the support has been the most common synthetic approach. However, since H2O2 formation occurs in a three-phase system, other models of distribution were investigated such as catalysts prepared with the active phase concentrated only on the surface layers of the support (egg-shell catalysts) or with a subsurface distribution (egg-white). Catalysts are prepared with different metal loading (0.1–10%) and through different deposition techniques, specifically: (i) wet impregnation; (ii) incipient wetness impregnation (or “dry” impregnation); (iii) deposition–precipitation.
Menegazzo et al. found [119] that the effect of gold addition to Pd in enhancing the yield of H2O2 is sensitive to the preparation method. In particular, the best catalytic results in terms of both productivity and selectivity were obtained co-impregnating Pd and Au on SiO2 rather than introducing the active components in sequence (first Au then Pd and vice versa). The HRTEM and TPR (Temperature Programmed Reductions) made it evident that the choice of the preparation method, i.e., incipient wetness (IW) co-impregnation rather than IW consecutive impregnations, is crucial for determining the morphology of the metallic active phase that strongly affects the catalytic performance in terms of both conversion and selectivity. The presence of a Pd-Au phase able to guarantee the availability of less energetic sites able to activate the oxygen molecule without dissociation can explain the catalytic performance of Pd-Au/SiO2 catalysts.
Sterchele et al. [184] investigated the effect of metal precursor reduction with different reducing agents on Pd-Au and Pd-Pt catalysts finding that formaldehyde, compared to hydrogen, could improve the activity and selectivity of Pd-Au and Pd-Pt catalysts.
Furthermore, acid and heat treatments on carbon supports can significantly improve the reaction performance of carbon supported catalysts. Hutchings and coworkers [57] found that, by using acid pretreated carbon materials, highly dispersed Au-Pd bimetallic catalysts could be prepared thus obtaining high H2O2 selectivity. Solsona et al. [98] also found that, by using acid pretreated orderly mesoporous carbon, the size of the Pd-Au alloy could be reduced to about 2 nm and the selectivity of H2O2 reached 99%. Moreover, Hutchings and coworkers [186,187] found that heat treatment (post-process) can improve the metal (Pd-Au) particle dispersion and morphology in carbon supported catalysts, thus improving their stability and recycling performance.
According to the same authors [54,56], the supported Au catalysts prepared by impregnation are more effective than the corresponding Au catalysts prepared by co-precipitation or deposition–precipitation. These authors also investigated [187] the preparation method and loading sequence of bimetallic catalysts finding that the Pd-Au/ZrO2 catalyst prepared first by Au deposition precipitation followed by Pd incipient wetness impregnation has a higher Pd content and better performance in H2O2 synthesis. They also reported that much higher H2O2 yields could be obtained with the non-calcined Pd-Au supported catalysts as compared with the corresponding calcined (at 400 °C) sample [52,54]. However, non-calcined catalysts are poorly stable towards leaching of both Au and Pd and cannot be reused successfully.
The same authors prepared Pd-Au bimetallic particles on both activated carbon and TiO2 supports with Pd{Au} (Pd-shell/Au-core) and Au{Pd} (Au-shell/Pd-core) morphologies by colloid immobilization technique [187]. It was found that, compared to the standard Pd-Au catalysts made by simultaneous addition and reduction of both metal precursors (in this case Au and Pd are randomly alloyed), both the Pd{Au} and Au{Pd} core–shell catalysts showed lower catalytic activity that could be caused by a high rate of H2O2 hydrogenation reaction.
Seo et al. investigated Pd@SiO2 core-shell catalysts observing a higher yield of hydrogen peroxide than traditionally supported Pd/SiO2 samples [183]. A volcano curve was observed for hydrogen peroxide production rate as a function of the shell thickness. When using catalysts with thin shells, the small exposed Pd surface area resulted in a lower hydrogen conversion and a lower hydrogen peroxide production rate. On the other hand, when the shell was thick, hydrogen conversion rate sharply decreased due to increased mass transfer resistance. Catalysts with intermediate shell thicknesses achieved high hydrogen conversion and hydrogen peroxide yield, owing to a better Pd specific surface area. The thinner the shell, the better for hydrogen peroxide synthesis, but a minimum thickness is necessary to achieve the maximum Pd specific surface area in the core-shell synthesis process.
Enhanced stability of the catalyst nanoparticles by adopting the core@shell structure is accompanied by restricted access of the reactant molecules to the Pd surface. The core@shell structure embeds the catalyst nanoparticle and the Pd area on which reactions can occur is reduced relative to the bare Pd catalyst. In order to overcome this limitation, a special type of a core@shell configuration, a core@void@shell structure, better known as a ‘yolk@shell,’ can be applied. Lee’s group prepared yolk@shell nanocatalysts using a method consisted of the following four steps: (i) Pd nanoparticle synthesis; (ii) a first encapsulation with SiO2 to prepare Pd@SiO2; (iii) a second encapsulation with ZrO2 to prepare Pd@SiO2@ZrO2; and (iv) etching of the silica layer from Pd@SiO2@ZrO2 with KOH solution [188]. Compared to Pd@SiO2, the as-synthesized Pd@void@ZrO2 catalyst has a 160% increase in the exposed specific Pd surface area and a remarkable 120% increase in hydrogen peroxide production.
Pd core–silica shell catalysts (PdX@SiO2, X = KBr, CTAB, SC) were prepared with and without the addition of three different functional molecules, potassium bromide (KBr), cetyltrimethyl ammonium bromide (CTAB) and sodium citrate dihydrate (SC), using a reverse micelle method [189]. The addition of functional molecules has a significant impact on the performance of the catalysts, and the H2O2 selectivity and productivity follow the order: PdKBr@SiO2 > PdCTAB@SiO2 > Pd@SiO2 > PdSC@SiO2. Among the prepared catalysts, the catalyst PdKBr@SiO2 had the highest H2O2 selectivity (98%) and productivity (716 mmol gPd−1 h−1). The reason for this was that the Br− of KBr can be easily selectively adsorbed on the coordinately unsaturated sites of the Pd particle surface, which could decrease the energy of the Pd surface and restrain side reactions by inhibiting the O–O bond cleavage of O2 and H2O2. Moreover, the catalyst PdKBr@SiO2 has better stability and recyclability than the conventional supported catalyst Pd/SiO2. TEM characterization shows that the addition of functional molecules can reduce the particle size of Pd particles, and the particle size of Pd follows the order: Pd >PdCTAB > PdKBr > PdSC.
Recently, ligand-modified palladium nanoparticles [190] capped with hexadecyl-2-hydroxyethyl-dimethylammonium dihydrogen phosphate (HHDMA) ligand and deposited on a carbon carrier probed useful to catalyze the direct synthesis of H2O2. The selectivity increases by increasing the amount of HHDMA on the surface, from 10% for naked nanoparticles up to 80% for modified nanoparticles. The catalyst remained stable over five consecutive reaction runs, owing to the high resistance towards leaching of the ligand because of its bond with the metal surface. DFT attributed this behavior to the adsorption mode of the reaction intermediates on the metal surface. According to the authors, whereas they lie flat in the absence of the organic shell, their electrostatic interaction with the ligand result in a unique vertical configuration, which prevents further dissociation and over-hydrogenation.
In 2018, a Pd/C catalyst was synthesized by selective adsorption deposition, in which a cationic palladium precursor such as [Pd(NH3)4]2+ was selectively adsorbed on a negatively charged activated carbon surface and subsequently slowly converted to palladium hydroxide with urea [191]. The formation of ultra-small and monodispersed Pd nanoparticles with a high Pd2+/Pd0 ratio resulted in high initial H2O2 productivity (8606 mmol H2O2/gPd h) and selectivity (95.1%). These values, under the same reaction conditions, were 12 and 7.1 times higher, respectively, than those of a Pd/C catalyst prepared by a conventional deposition–precipitation method.
Hu et al. [192] found that, in comparison with other carbon supports, graphitized carbon support can improve the selectivity to H2O2, and the authors attributed that to the high degree of structural order graphitized.
Other less traditional methods have been used. Mori et al. [193,194] reported that the activity of Pd-Au/Ti-MCM-41 and nanoscale Pd/CsHPA/SiO2 (CsHPA: heteropoly acid cesium salt) catalysts prepared by light deposition with a high pressure mercury lamp was much better than that prepared by simple deposition. The authors thought that, under illumination, optical excitation sites of the support materials can interact with metal precursors to form smaller metal nanoparticles.
Howe et al. [195] used a one pot microwave synthesis of highly stable AuPd@Pd supported nanoparticles. These particles have a core–shell structure and can be quickly prepared. The catalysts probed maintaining activity for four reaction cycles.
Pd catalysts supported on TiO2 and SiO2 were prepared by a sonochemical method [196]. By this technique, a homogeneous and small size nucleation is often reported, and oxygen vacancies in TiO2 are reported to be strong adsorption sites for Pd nucleation. This synergistic effect resulted in a high Pd surface area for TiO2 supported sample, owing to small particle nucleation at oxygen vacancies. On the contrary, sonochemically prepared Pd/SiO2 showed a very low Pd dispersion and a large Pd particle size. In the direct synthesis of H2O2, sonochemical Pd/TiO2 demonstrated a quite high yield (16%) but a reduction in H2O2 selectivity due to electron exchange between Pd and TiO2, which led to an electron-rich state of Pd, as explained by XPS.
A hierarchically porous Pd/SiO2 catalyst was synthesized by combination of miniemulsion polymerisation in water to prepare a Pd-containing latex template and sol-gel synthesis in controlled conditions to tailor the silica porosity [197]. The method consists (Figure 10) in the preparation of a styrene miniemulsion containing an organometallic Pd-precursor soluble within the monomer droplets. Then, the emulsion polymerization of styrene takes place, leading to the transformation of the monomer droplets into Pd-containing polymer particles. Subsequently, this hybrid Pd-polystyrene latex is used as template for the sol-gel synthesis under controlled conditions to modulate the final silica matrix porosity. After removal of the polystyrene template, the obtained material presented a hierarchical porosity (micro-, meso- and macropores) and an average surface area of 711 m2g−1.
This novel synthetic protocol allowed the preparation of silica-supported Pd catalysts, controlling the support hierarchical porosity and the preferential location of the Pd nanoparticles in the macropores. This material was then evaluated in the direct synthesis of H2O2, either after calcination or after calcination and reduction pre-treatment, exhibiting structural resistance at the reaction conditions and a stable selectivity (46%). No mass transfer limitations inside the catalyst particles were observed in a slurry batch reactor as long as the particles remain under 30 μm. This material with hierarchical porosity could potentially be used in larger particles in e.g., fixed bed reactors or in thick coatings of Pd/SiO2 on structured catalysts such as monoliths, for a continuous operation.
10. Conclusions
As is clear from the foregoing, the “dream catalyst” capable of conjugating industrially relevant H2O2 productivities with highly efficient hydrogen consumption, stability on stream, low energy input and safe reaction conditions has not yet been invented. Rather, from the analysis of the different features characterizing the current catalysts, some trends emerge more or less clearly so that we can at least try to define the essential properties that a catalyst should possess to move close to “dream”.
Pd is undoubtedly the essential component; however, in order to depress its ability to trigger hydrogen peroxide decomposition/hydrogenation and increase its selectivity, Pd should exist in a partially oxidized electronic configuration, generally achieved through alloying with other metals the most prominent ones being Au or Pt. Another essential feature to maximize H2O2 selectivity is the Pd or Pd-M particle size. This should be neither too large because this would result in depressing the overall catalytic activity, nor too small to avoid an excess of highly unsaturated active sites that would boost water formation via dissociative dioxygen chemisorption. In this respect, the use of additives such as halides (otherwise undesired for practical applications) or better N-containing ligands seem to be a good practice to poison the highly unsaturated sites and in the latter case also stabilize metal particles against sintering, leaching or physical detachment. Unfortunately, as has been seen in Section 9, how to achieve all that in a single catalyst is still unclear since, as far as the preparation method is concerned, there are many ideas on the ground but none seems to outperform the other ones. Core–shell catalysts seem to be promising, but they are rather complex to prepare a feature that does not encourage industrial applications where simplicity and reliability of preparation are essential properties for an industrial catalyst.
The support should have proper acidity and/or a low isoelectric point to stabilize the newly formed H2O2 molecule, as well as a suitable textural property to impart high mechanical and chemical stability. An acid-pretreated carbon appears to be the best choice when operating in organic solvents such as methanol, a medium that would be suitable for post synthesis solution concentration and for a wide variety of applications in organic synthesis. On the other hand, a Pd-Au exchanging a Cs-containing heteropolyacid catalyst seems to be the current benchmark for water as the solvent [107]. A proper formulation of the support, i.e., an acid pre-treatment of the sample or already incorporated acidic functions, will avoid halides and/or inorganic acids in the reaction medium to increase selectivity.
From these considerations and the above overview on the state of the art in this area, it may seem that the “dream catalyst” is at hand. Unfortunately, in our opinion, this is a wrong impression since the distance to be covered is still long. In fact, it has to be pointed out that the catalysts capable of approaching industrially interesting H2O2 productivities (at least 5% w/v concentrations) with acceptable selectivity (at least 70%) and stable on time are actually very few in a myriad of different catalysts prepared and tested in the past twenty years. The vast majority of these have productivities that hardly reach 1%, or they show 90% selectivity but at very low conversions and poor productivity, or they have relatively high productivity and selectivity but rapidly declining with time. After one century from the first report and at least twenty years of intense research in the area by academia, this general situation raises the legitimate question of whether the “dream catalyst” for the direct synthesis of hydrogen peroxide will ever be discovered or whether non catalytic approaches such as e.g., through electrochemical devices [198] or using plasma techniques [199] will be more successful.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Myers, R.L. The 100 Most Important Chemical Compounds: A Reference Guide; Greenwood Publishing Group: Westport, CT, USA, 2007. [Google Scholar]
- Hydrogen Peroxide Market Price. Available online: https://www.alibaba.com/showroom/hydrogen-peroxide-market-price.html (accessed on 7 March 2019).
- Lewis, R.J.; Hutchings, G.J. Recent Advances in the Direct Synthesis of H2O2. ChemCatChem 2019, 11, 298–308. [Google Scholar] [CrossRef]
- Henkel, H.; Weber, W. Manufacture of Hydrogen Peroxide. U.S. Patent 1,108,752, 25 August 1914. [Google Scholar]
- Li, J.; Yoshizawa, K. Mechanistic aspects in the direct synthesis of hydrogen peroxide on PdAu catalyst from first principles. Catal. Today 2015, 248, 142–148. [Google Scholar] [CrossRef]
- García-Serna, J.; Moreno, T.; Biasi, P.; Cocero, M.J.; Mikkola, J.P.; Salmi, T. Engineering in direct synthesis of hydrogen peroxide: Targets, reactors and guidelines for operational conditions. Green Chem. 2014, 16, 2320–2343. [Google Scholar] [CrossRef]
- Piqueras, C.M.; García-Serna, J.; Cocero, M.J. Estimation of lower flammability limits in high-pressure systems. Application to the direct synthesis of hydrogen peroxide using supercritical and near-critical CO2 and air as diluents. J. Supercrit. Fluids 2011, 56, 33–40. [Google Scholar] [CrossRef]
- Biasi, P.; Menegazzo, F.; Canu, P.; Pinna, F.; Salmi, T. Role of a Functionalized Polymer (K2621) and an Inorganic Material (Sulphated Zirconia) as Supports in Hydrogen Peroxide Direct Synthesis in a Continuous Reactor. Ind. Eng. Chem. Res. 2013, 52, 15472–15480. [Google Scholar] [CrossRef]
- Seo, M.; Kim, J.H.; Han, S.; Lee, K. Direct Synthesis of Hydrogen Peroxide from Hydrogen and Oxygen Using Tailored Pd Nanocatalysts: A Review of Recent Findings. Catal. Surv. Asia 2017, 21, 1–12. [Google Scholar] [CrossRef]
- Yi, Y.; Wang, L.; Li, G.; Guo, H. Review on research progress in the direct synthesis of hydrogen peroxide from hydrogen and oxygen: Noble-metal catalytic method, fuel-cell method and plasma method. Catal. Sci. Technol. 2016, 6, 1593–1610. [Google Scholar] [CrossRef]
- Edwards, J.K.; Freakley, S.J.; Lewis, R.J.; Pritchard, J.C.; Hutchings, G.J. Advances in the direct synthesis of hydrogen peroxide from hydrogen and oxygen. Catal. Today 2015, 248, 3–9. [Google Scholar] [CrossRef]
- Dittmeyer, R.; Grunwaldt, J.D.; Pashkova, A. A review of catalyst performance and novel reaction engineering concepts in direct synthesis of hydrogen peroxide. Catal. Today 2015, 248, 149–159. [Google Scholar] [CrossRef]
- Campos-Martin, J.M.; Blanco-Brieva, G.; Fierro, J.L.G. Hydrogen Peroxide Synthesis: An Outlook beyond the Anthraquinone Process. Angew. Chem. Int. Ed. 2006, 45, 6962–6984. [Google Scholar] [CrossRef] [PubMed]
- Hooper, G.W. Catalytic Production of Hydrogen Peroxide from Its Elements. U.S. Patent 3,336,112, 15 August 1967. [Google Scholar]
- Izumi, Y.; Miyazaki, H.; Kawahara, I. Process for the Direct Production of Hydrogen Peroxide from Hydrogen and Oxygen. DE Patent 2,528,601 A1, 1976. [Google Scholar]
- Gosser, L.W.; Schwartz, J.A.T. Catalytic Process for Making Hydrogen Peroxide from Hydrogen and Oxygen Employing a Bromide Promoter. U.S. Patent 4,772,458, 1988. [Google Scholar]
- Wang, F.; Xia, C.; De Visser, S.P.; Wang, Y. How Does the Oxidation State of Palladium Surfaces Affect the Reactivity and Selectivity of Direct Synthesis of Hydrogen Peroxide from Hydrogen and Oxygen Gases? A Density Functional Study. J. Am. Chem. Soc. 2019, 141, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Pospelova, T.A.; Kobozev, N.I. Palladium catalyzed synthesis of hydrogen peroxide from its elements II: Active centers of palladium. Rus. J. Phys. Chem. 1961, 35, 262. [Google Scholar]
- Burch, R.; Ellis, P.R. An investigation of alternative catalytic approaches for the direct synthesis of hydrogen peroxide from hydrogen and oxygen. Appl. Catal. B 2003, 42, 203–211. [Google Scholar] [CrossRef]
- Gaikwad, A.G.; Sansare, S.D.; Choudhary, V.R. Direct oxidation of hydrogen to hydrogen peroxide over Pd-containing fluorinated or sulfated Al2O3, ZrO2, CeO2, ThO2, Y2O3 and Ga2O3 catalysts in stirred slurry reactor at ambient conditions. J. Mol. Catal. A Chem. 2002, 181, 143–149. [Google Scholar] [CrossRef]
- Paredes Olivera, P.; Patrito, E.M.; Sellers, H. Hydrogen peroxide synthesis over metallic catalysts. Surf. Sci. 1994, 313, 25–40. [Google Scholar] [CrossRef]
- Jones, C.A.; Grey, R.A. Process for Producing Hydrogen Peroxide. U.S. Patent 6,468,496, 22 October 2002. [Google Scholar]
- Landon, P.; Collier, P.J.; Papworth, A.J.; Kiely, C.J.; Hutchings, G.J. Direct formation of hydrogen peroxide from H2/O2 using a gold catalyst. Chem. Commun. 2002, 2058–2059. [Google Scholar] [CrossRef]
- Wells, D.H., Jr.; Delgass, W.N.; Thomson, K.T. Formation of hydrogen peroxide from H2 and O2 over a neutral gold trimer: A DFT study. J. Catal. 2004, 225, 69–77. [Google Scholar] [CrossRef]
- Fernandez, E.M.; Ordejón, P.; Balbás, L.C. Theoretical study of O2 and CO adsorption on Au nclusters (n = 5–10). Chem. Phys. Lett. 2005, 408, 252–257. [Google Scholar] [CrossRef]
- Wells, D.H., Jr.; Delgass, W.N.; Thomson, K.T. Density functional theory investigation of gold cluster geometry and gas-phase reactivity with O2. J. Chem. Phys. 2002, 117, 10597. [Google Scholar] [CrossRef]
- Phala, N.S.; Klatt, G.; van Steen, E. A DFT study of hydrogen and carbon monoxide chemisorption onto small gold clusters. Chem. Phys. Lett. 2004, 395, 33–37. [Google Scholar] [CrossRef]
- Okumura, M.; Kitagawa, Y.; Haruta, M.; Yamaguchi, K. The interaction of neutral and charged Au clusters with O2, CO and H2. Appl. Catal. A 2005, 291, 37–44. [Google Scholar] [CrossRef]
- Okumura, M.; Kitagawa, Y.; Yamagchuhi, K.; Akita, T.; Tsubota, S.; Haruta, M. Direct Production of Hydrogen Peroxide from H2 and O2 over Highly Dispersed Au catalysts. Chem. Lett. 2003, 32, 822–823. [Google Scholar] [CrossRef]
- Ishihara, T.; Ohura, Y.; Yoshida, S.; Hata, Y.; Nishiguchi, H.; Takita, Y. Synthesis of hydrogen peroxide by direct oxidation of H2 with O2 on Au/SiO2 catalyst. Appl. Catal. A 2005, 291, 215–221. [Google Scholar] [CrossRef]
- Choudhary, V.R.; Sansare, S.D.; Gaikwad, A.G. Direct Oxidation of H2 to H2O2 and Decomposition of H2O2 Over Oxidized and Reduced Pd-Containing Zeolite Catalysts in Acidic Medium. Catal. Lett. 2002, 84, 81–87. [Google Scholar] [CrossRef]
- Chinta, S.; Lunsford, J.H. A mechanistic study of H2O2 and H2O formation from H2 and O2 catalyzed by palladium in an aqueous medium. J. Catal. 2004, 225, 249–255. [Google Scholar] [CrossRef]
- Melada, S.; Rioda, R.; Menegazzo, F.; Pinna, F.; Strukul, G. Direct synthesis of hydrogen peroxide on zirconia-supported catalysts under mild conditions. J. Catal. 2006, 239, 422–430. [Google Scholar] [CrossRef]
- Lunsford, J.H. The direct formation of H2O2 from H2 and O2 over palladium catalysts. J. Catal. 2003, 216, 455–460. [Google Scholar] [CrossRef]
- Abate, S.; Centi, G.; Perathoner, S.; Melada, S.; Pinna, F.; Strukul, G. The issue of selectivity in the direct synthesis of H2O2 from H2 and O2: The role of the catalyst in relation to the kinetics of reaction. Top. Catal. 2006, 38, 181–193. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S.; Abate, S. Modern Heterogeneous Oxidation Catalysis; Wiley-VCH Verlag GmbH & Co. KgaA: Weinheim, Germany, 2009; p. 253. [Google Scholar]
- Han, R.Y.; Lunsford, J.H. Direct formation of H2O2 from H2 and O2 over a Pd/SiO2 catalyst: The roles of the acid and the liquid phase. J. Catal. 2005, 230, 313–316. [Google Scholar] [CrossRef]
- Choudary, V.R.; Samanta, C.; Choudhary, T.V. Influence of nature/concentration of halide promoters and oxidation state on the direct oxidation of H2 to H2O2 over Pd/ZrO2 catalysts in aqueous acidic medium. Catal. Commun. 2007, 8, 1310–1316. [Google Scholar] [CrossRef]
- Rodriguez, J.A. The chemical properties of bimetallic surfaces: Importance of ensemble and electronic effects in the adsorption of sulfur and SO2. Prog. Surf. Sci. 2006, 81, 141–189. [Google Scholar] [CrossRef]
- Li, G.; Edwards, J.K.; Carley, A.F.; Hutchings, G.J. Direct synthesis of hydrogen peroxide from H2 and O2 and in situ oxidation using zeolite-supported catalysts. Catal. Commun. 2007, 8, 247–250. [Google Scholar] [CrossRef]
- Menegazzo, F.; Burti, P.; Signoretto, M.; Manzoli, M.; Vankova, S.; Boccuzzi, F.; Pinna, F.; Strukul, G. Effect of the addition of Au in zirconia and ceria supported Pd catalysts for the direct synthesis of hydrogen peroxide. J. Catal. 2008, 257, 369–381. [Google Scholar] [CrossRef]
- Landon, P.; Collier, P.J.; Carley, A.F.; Chadwick, D.; Papworth, A.J.; Burrows, A.; Kiely, C.J.; Hutchings, G.J. Direct synthesis of hydrogen peroxide from H2 and O2 using Pd and Au catalysts. Phys. Chem. Chem. Phys. 2003, 5, 1917–1923. [Google Scholar] [CrossRef]
- Li, G.; Edwards, J.; Carley, A.F.; Hutchings, G.J. Direct synthesis of hydrogen peroxide from H2 and O2 using zeolite-supported Au-Pd catalysts. Catal. Today 2007, 122, 361–364. [Google Scholar] [CrossRef]
- Li, G.; Edwards, J.; Carley, A.F.; Hutchings, G.J. Direct synthesis of hydrogen peroxide from H2 and O2 using zeolite-supported Au catalysts. Catal. Today 2006, 114, 369–371. [Google Scholar] [CrossRef]
- Solsona, B.E.; Edwards, J.K.; Landon, P.; Carley, A.F.; Herzing, A.; Kiely, C.J.; Hutchings, G.J. Direct Synthesis of Hydrogen Peroxide from H2 and O2 Using Al2O3 Supported Au−Pd Catalysts. Chem. Mater. 2006, 18, 2689–2695. [Google Scholar] [CrossRef]
- Menegazzo, F.; Signoretto, M.; Manzoli, M.; Boccuzzi, F.; Cruciani, G.; Pinna, F.; Strukul, G. Influence of the preparation method on the morphological and composition properties of Pd–Au/ZrO2 catalysts and their effect on the direct synthesis of hydrogen peroxide from hydrogen and oxygen. J. Catal. 2009, 268, 122–130. [Google Scholar] [CrossRef]
- Deguchi, T.; Yamano, H.; Takenouchi, S.; Iwamoto, M. Enhancement of catalytic activity of Pd-PVP colloid for direct H2O2 synthesis from H2 and O2 in water with addition of 0.5 atom% Pt or Ir. Catal. Sci. Technol. 2018, 8, 1002–1015. [Google Scholar] [CrossRef]
- Choudhary, V.R.; Gaikwad, A.G.; Sansare, S.D. Nonhazardous Direct Oxidation of Hydrogen to Hydrogen Peroxide Using a Novel Membrane Catalyst. Angew. Chem. Int. Ed. 2001, 40, 1776–1779. [Google Scholar] [CrossRef]
- Abate, S.; Melada, S.; Centi, G.; Perathoner, S.; Pinna, F.; Strukul, G. Performances of Pd-Me (Me = Ag, Pt) catalysts in the direct synthesis of H2O2 on catalytic membranes. Catal. Today 2006, 117, 193–198. [Google Scholar] [CrossRef]
- Liu, Q.; Bauer, J.C.; Schaak, R.E.; Lunsford, J.H. Direct synthesis of H2O2 from H2 and O2 over Pd–Pt/SiO2 bimetallic catalysts in a H2SO4/ethanol system. Appl. Catal. A 2008, 339, 130–136. [Google Scholar] [CrossRef]
- Choudhary, V.R.; Samanta, C.; Choudhary, T.V. Direct oxidation of H2 to H2O2 over Pd-based catalysts: Influence of oxidation state, support and metal additives. Appl. Catal. A 2006, 308, 128–133. [Google Scholar] [CrossRef]
- Ntainjua, E.N.; Freakley, S.J.; Hutchings, G.J. Direct Synthesis of Hydrogen Peroxide Using Ruthenium Catalysts. Top. Catal. 2012, 55, 718–722. [Google Scholar] [CrossRef]
- Gu, J.; Wang, S.; He, Z.; Han, Y.; Zhang, J. Direct synthesis of hydrogen peroxide from hydrogen and oxygen over activated-carbon supported Pd–Ag alloy catalysts. Catal. Sci. Technol. 2016, 6, 809–817. [Google Scholar] [CrossRef]
- Edwards, J.K.; Solsona, B.E.; Landon, P.; Carley, A.F.; Herzing, A.; Kiely, C.J.; Hutchings, G.J. Direct synthesis of hydrogen peroxide from H2 and O2 using TiO2-supported Au-Pd catalysts. J. Catal. 2005, 236, 69–79. [Google Scholar] [CrossRef]
- Freakley, S.J.; Piccinini, M.; Edwards, J.K.; Ntainjua, E.N.; Moulijn, J.A.; Hutchings, G.J. Effect of Reaction Conditions on the Direct Synthesis of Hydrogen Peroxide with a AuPd/TiO2 Catalyst in a Flow Reactor. ACS Catal. 2013, 3, 487–501. [Google Scholar] [CrossRef]
- Edwards, J.K.; Solsona, B.; Landon, P.; Carley, A.F.; Herzing, A.; Watanabe, M.; Kiely, C.J.; Hutchings, G.J. Direct synthesis of hydrogen peroxide from H2 and O2 using Au-Pd/Fe2O3 catalysts. J. Mater. Chem. 2005, 15, 4595–4600. [Google Scholar] [CrossRef]
- Edwards, J.K.; Solsona, B.; Ntainjua, E.; Carley, A.F.; Herzing, A.A.; Kiely, C.J.; Hutchings, G.J. Switching Off Hydrogen Peroxide Hydrogenation in the Direct Synthesis Process. Science 2009, 323, 1037–1041. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.K.; Pritchard, J.; Piccinini, M.; Shaw, G.; He, Q.; Carley, A.F.; Kiely, C.J.; Hutchings, G.J. The effect of heat treatment on the performance and structure of carbon-supported Au–Pd catalysts for the direct synthesis of hydrogen peroxide. J. Catal. 2012, 292, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Kumar, D.; Yi, C.W.; Goodman, D.W. The promotional effect of gold in catalysis by palladium-gold. Science 2005, 310, 291–293. [Google Scholar] [CrossRef] [PubMed]
- Nomura, Y.; Ishihara, T.; Hata, Y.; Kitawaki, K.; Kaneko, K.; Matsumoto, H. Nanocolloidal Pd-Au as Catalyst for the Direct Synthesis of Hydrogen Peroxide from H2 and O2. ChemSusChem 2008, 1, 619–621. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Staykov, A.; Ishihara, T.; Yoshizawa, K. Theoretical Study of the Decomposition and Hydrogenation of H2O2 on Pd and Au@Pd Surfaces: Understanding toward High Selectivity of H2O2 Synthesis. J. Phys. Chem. C 2011, 115, 7392–7398. [Google Scholar] [CrossRef]
- Li, J.; Ishihara, T.; Yoshizawa, K. Theoretical Revisit of the Direct Synthesis of H2O2 on Pd and Au@Pd Surfaces: A Comprehensive Mechanistic Study. J. Phys. Chem. C 2011, 115, 25359–25367. [Google Scholar] [CrossRef]
- Sterchele, S.; Biasi, P.; Centomo, P.; Canton, P.; Campestrini, S.; Salmi, T.; Zecca, M. Pd-Au and Pd-Pt catalysts for the direct synthesis of hydrogen peroxide in absence of selectivity enhancers. Appl. Catal. A 2013, 46, 160–164. [Google Scholar] [CrossRef]
- Ouyang, L.; Da, G.J.; Tian, P.F.; Chen, T.; Liang, G.; Xu, J.; Han, Y. Insight into active sites of Pd–Au/TiO2 catalysts in hydrogen peroxide synthesis directly from H2 and O2. J. Catal. 2014, 311, 129–136. [Google Scholar] [CrossRef]
- Staykov, A.; Kamachi, T.; Ishihara, T.; Yoshizawa, K. Theoretical study of the direct synthesis of H2O2 on Pd and Pd/Au surfaces. J. Phys. Chem. C 2008, 112, 19501–19505. [Google Scholar] [CrossRef]
- Ham, H.C.; Hwang, G.S.; Han, J.; Nam, S.W.; Lim, T.H. On the Role of Pd Ensembles in Selective H2O2 Formation on PdAu Alloys. J. Phys. Chem. C 2009, 113, 12943–12945. [Google Scholar] [CrossRef]
- Han, Y.F.; Zhong, Z.Y.; Ramesh, K.; Chen, F.; Chen, L.; White, T.; Tay, Q.; Yaakub, S.N.; Wang, Z. Au promotional effects on the synthesis of H2O2 directly from H2 and O2 on supported Pd−Au alloy catalysts. J. Phys. Chem. C 2007, 111, 8410–8413. [Google Scholar] [CrossRef]
- Gudarzi, D.; Ratchananusorn, W.; Turunen, I.; Heinonen, M.; Salmi, T. Promotional effects of Au in Pd–Au bimetallic catalysts supported on activated carbon cloth (ACC) for direct synthesis of H2O2 from H2 and O2. Catal. Today 2015, 248, 58–68. [Google Scholar] [CrossRef]
- Kanungo, S.; van Haandel, L.; Hensen, E.J.M.; Schouten, J.C.; d’Angelo, M.F.N. Direct synthesis of H2O2 in AuPd coated micro channels: An in-situ X-Ray absorption spectroscopic study. J. Catal. 2019, 370, 200–209. [Google Scholar] [CrossRef]
- Xu, J.; Ouyang, L.; Da, G.J.; Song, Q.Q.; Yang, X.J.; Han, Y.F. Pt promotional effects on Pd–Pt alloy catalysts for hydrogen peroxide synthesis directly from hydrogen and oxygen. J. Catal. 2012, 285, 74–82. [Google Scholar] [CrossRef]
- Quon, S.; Jo, D.Y.; Han, G.; Han, S.S.; Seo, M.; Lee, K. Role of Pt atoms on Pd(1 1 1) surface in the direct synthesis of hydrogen peroxide: Nano-catalytic experiments and DFT calculations. J. Catal. 2018, 368, 237–247. [Google Scholar] [CrossRef]
- Meiers, R.; Holderich, W.F. Epoxidation of propylene and direct synthesis of hydrogen peroxide by hydrogen and oxygen. Catal. Lett. 1999, 59, 161–163. [Google Scholar] [CrossRef]
- Meiers, R.; Dingerdissen, U.; Holderich, W.F. Synthesis of propylene oxide from propylene, oxygen and hydrogen catalyzed by palladium–platinum–containing titanium silicalite. J. Catal. 1998, 176, 376–386. [Google Scholar] [CrossRef]
- Monnier, J.R. The direct epoxidation of higher olefins using molecular oxygen. Appl. Catal. A 2001, 221, 73–91. [Google Scholar] [CrossRef]
- Melada, S.; Pinna, F.; Strukul, G.; Perathoner, S.; Centi, G. Direct synthesis of H2O2 on monometallic and bimetallic catalytic membranes using methanol as reaction medium. J. Catal. 2006, 237, 213–219. [Google Scholar] [CrossRef]
- Bernardotto, G.; Menegazzo, F.; Pinna, F.; Signoretto, M.; Cruciani, G.; Strukul, G. New Pd–Pt and Pd–Au catalysts for an efficient synthesis of H2O2 from H2 and O2 under very mild conditions. Appl. Catal. A 2009, 358, 129–135. [Google Scholar] [CrossRef]
- United States Environmental Protection Agency. Available online: https://www.epa.gov/greenchemistry/presidential-green-chemistry-challenge-2007-greener-reaction-conditions-award (accessed on 7 March 2019).
- Edwards, J.K.; Pritchard, J.; Miedziak, P.J.; Piccinini, M.; Carley, A.F.; He, Q.; Kiely, C.J.; Hutchings, G.J. The direct synthesis of hydrogen peroxide using platinum promoted gold–palladium catalysts. Catal. Sci. Technol. 2014, 4, 3244–3250. [Google Scholar] [CrossRef]
- Edwards, J.K.; Pritchard, J.; Lu, L.; Piccinini, M.; Shaw, G.; Carley, A.F.; Morgan, D.J.; Kiely, C.J.; Hutchings, G.J. The direct synthesis of hydrogen peroxide using platinum-promoted gold-palladium catalysts. Angew. Chem. Int. Ed. 2014, 53, 2381–2384. [Google Scholar] [CrossRef] [PubMed]
- Maity, S.; Eswaramoorthy, M. Ni-Pd bimetallic catalysts for the direct synthesis of H2O2-unusual enhancement of Pd activity in the presence of Ni. J. Mater. Chem. A 2016, 4, 3233–3237. [Google Scholar] [CrossRef]
- Xu, H.; Cheng, D.; Gao, Y. Design of High-Performance Pd-Based Alloy Nanocatalysts for Direct Synthesis of H2O2. ACS Catal. 2017, 7, 2164–2170. [Google Scholar] [CrossRef]
- Sun, M.; Liu, X.; Hang, B. Dynamically self-activated catalyst for direct synthesis of hydrogen peroxide (H2O2). Mater. Today Energy 2018, 10, 307–316. [Google Scholar] [CrossRef]
- Wang, S.; Gao, K.; Li, W.; Zhang, J. Effect of Zn addition on the direct synthesis of hydrogen peroxide over supported palladium catalysts. Appl. Catal. A 2017, 531, 89–95. [Google Scholar] [CrossRef]
- Gong, X.; Yang, Z.; Peng, L.; Zhou, A.; Liu, Y.; Liu, Y. In-situ synthesis of hydrogen peroxide in a novel Zn-CNTs-O2 system. J. Power Sources 2018, 378, 190–197. [Google Scholar] [CrossRef]
- Wilson, N.M.; Schröder, J.; Priyadarshini, P.; Bregante, D.T.; Kunz, S.; Flaherty, D.W. Direct synthesis of H2O2 on PdZn nanoparticles: The impact of electronic modifications and heterogeneity of active sites. J. Catal. 2018, 368, 261–274. [Google Scholar] [CrossRef]
- Tian, P.; Xu, X.; Ao, C.; Ding, D.; Li, W.; Si, R.; Tu, W.; Xu, J.; Han, Y. Direct and Selective Synthesis of Hydrogen Peroxide over Palladium–Tellurium Catalysts at Ambient Pressure. ChemSusChem 2017, 10, 3342–3346. [Google Scholar] [CrossRef] [PubMed]
- Freakley, S.J.; He, Q.; Harrhy, J.H.; Lu, L.; Crole, D.A.; Morgan, D.J.; Ntainjua, E.N.; Edwards, J.K.; Carley, A.F.; Borisevich, A.Y.; et al. Palladium-tin catalysts for the direct synthesis of H2O2 with high selectivity. Science 2016, 351, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Xu, X.; Tian, P.; Liu, X.; Xu, J.; Han, Y. Promotional effects of Sb on Pd-based catalysts for the direct synthesis of hydrogen peroxide at ambient pressure. Chin. J. Catal. 2018, 39, 673–681. [Google Scholar] [CrossRef]
- Nugraha, M.; Tsai, M.; Rick, J.; Su, W.; Chou, H.; Hwang, B.J. DFT study reveals geometric and electronic synergisms of palladium-mercury alloy catalyst used for hydrogen peroxide formation. Appl. Catal. A 2017, 547, 69–74. [Google Scholar] [CrossRef]
- Gosser, L.W.; Paoli, M.A. Method for Catalytic Production of Hydrogen Peroxide. U.S. Patent 5,135,731, 4 August 1992. [Google Scholar]
- Dalton, A.I., Jr.; Skinner, R.W. Synthesis of Hydrogen Peroxide. U.S. Patent 4,336,239, 22 June 1982. [Google Scholar]
- Liu, Q.; Gath, K.; Bauer, J.C.; Schaak, R.E.; Lunsford, J.H. The Active Phase in the Direct Synthesis of H2O2 from H2 and O2 over Pd/SiO2 catalyst in a H2SO4/Ethanol System. Catal. Lett. 2009, 132, 342–348. [Google Scholar] [CrossRef]
- Menegazzo, F.; Signoretto, M.; Frison, G.; Pinna, F.; Strukul, G.; Manzoli, M.; Boccuzzi, F. When high metal dispersion has a detrimental effect: Hydrogen peroxide direct synthesis under very mild and nonexplosive conditions catalyzed by Pd supported on silica. J. Catal. 2012, 290, 143–150. [Google Scholar] [CrossRef]
- Biasi, P.; Canu, P.; Menegazzo, F.; Pinna, F.; Salmi, T. Direct Synthesis of Hydrogen Peroxide in a Trickle Bed Reactor: Comparison of Pd-Based Catalysts. Ind. Eng. Chem. Res. 2012, 51, 8883–8890. [Google Scholar] [CrossRef] [Green Version]
- Edwards, J.K.; Thomas, A.; Solsona, B.; Landon, P.; Carley, A.F.; Hutchings, G. Comparison of supports for the direct synthesis of hydrogen peroxide from H2 and O2 using Au–Pd catalysts. Catal. Today 2007, 122, 397–402. [Google Scholar] [CrossRef]
- Yook, S.; Kwon, H.C.; Kim, Y.; Choi, W.; Choi, M. Significant roles of carbon pore and surface structure in AuPd/C catalyst for achieving high chemoselectivity in direct hydrogen peroxide synthesis. ACS Sustain. Chem. Eng. 2017, 5, 1208–1216. [Google Scholar] [CrossRef]
- Lee, J.W.; Kim, J.K.; Kang, T.H.; Lee, E.J.; Song, I.K. Direct synthesis of hydrogen peroxide from hydrogen and oxygen over palladium catalyst supported on heteropolyacid-containing ordered mesoporous carbon. Catal. Today 2017, 293–394, 49–55. [Google Scholar] [CrossRef]
- García, T.T.; Agouram, S.; Dejoz, A.; Sánchez-Royo, J.F.; Torrente-Murciano, L.; Solsona, B. Enhanced H2O2 production over Au-rich bimetallic Au–Pd nanoparticles on ordered mesoporous carbons. Catal. Today 2015, 248, 48–57. [Google Scholar] [CrossRef]
- Park, S.; Jung, J.; Seo, J.; Kim, T.; Chung, Y.; Oh, S.; Song, I. Direct synthesis of hydrogen peroxide from hydrogen and oxygen over palladium catalysts supported on SO3H-Functionalized SiO2 and TiO2. Catal. Lett. 2009, 130, 604–607. [Google Scholar] [CrossRef]
- Wu, M.; Sápi, A.; Avila, A.; Szabó, M.; Hiltunen, J.; Huuhtanen, M.; Tóth, G.; Kukovecz, Á.; Kónya, Z.; Keiski, R.; et al. Enhanced photocatalytic activity of TiO2 nanofibers and their flexible composite films: Decomposition of organic dyes and efficient H2 generation from ethanol–water mixtures. Nano Res. 2011, 4, 360–369. [Google Scholar] [CrossRef]
- Ghedini, E.; Menegazzo, F.; Signoretto, M.; Manzoli, M.; Pinna, F.; Strukul, G. Mesoporous silica as supports for Pd-catalyzed H2O2 direct synthesis: Effect of the textural properties of the support on the activity and selectivity. J. Catal. 2010, 273, 266–273. [Google Scholar] [CrossRef]
- Rodríguez-Gómez, A.; Platero, F.; Caballero, A.; Colón, G. Improving the direct synthesis of hydrogen peroxide from hydrogen and oxygen over Au-Pd/SBA-15 catalysts by selective functionalization. Mol. Catal. 2018, 445, 142–151. [Google Scholar] [CrossRef]
- Bernardini, A.; Gemo, N.; Biasi, P.; Canu, P.; Mikkola, J.P.; Salmi, T.; Lanza, R. Direct synthesis of H2O2 over Pd supported on rare earths promoted zirconia. Catal. Today 2015, 256, 294–301. [Google Scholar] [CrossRef]
- Park, S.Y.; Lee, J.G.; Song, J.H.; Kim, T.J.; Chung, Y.M.; Oh, S.H.; Song, I.K. Direct synthesis of hydrogen peroxide from hydrogen and oxygen over Pd/HZSM-5 catalysts: Effect of Brönsted acidity. J. Mol. Catal. A Chem. 2012, 363–364, 230–236. [Google Scholar] [CrossRef]
- Park, S.Y.; Park, D.R.; Choi, J.H.; Kim, T.J.; Chung, Y.M.; Oh, S.H.; Song, I.K. Direct synthesis of hydrogen peroxide from hydrogen and oxygen over insoluble Cs2.5H0.5PW12O40 heteropolyacid supported on Pd/MCF. J. Mol. Catal. A Chem. 2010, 332, 76–83. [Google Scholar] [CrossRef]
- Park, S.Y.; Choi, J.H.; Kim, T.J.; Chung, Y.M.; Oh, S.H.; Song, I.K. Direct synthesis of hydrogen peroxide from hydrogen and oxygen over Pd/CsXH3−XPW12O40/MCF (X = 1.7, 2.0, 2.2, 2.5, and 2.7) catalysts. J. Mol. Catal. A Chem. 2012, 353–354, 37–43. [Google Scholar] [CrossRef]
- Ntainjua, E.N.; Piccinini, M.; Freakley, S.J.; Pritchard, J.C.; Edwards, J.K.; Carley, A.F.; Hutchings, G.J. Direct synthesis of hydrogen peroxide using Au–Pd-exchanged and supported heteropolyacid catalysts at ambient temperature using water as solvent. Green Chem. 2012, 14, 170–181. [Google Scholar] [CrossRef]
- Park, S.Y.; Park, D.R.; Choi, J.H.; Kim, T.J.; Chung, Y.M.; Oh, S.; Song, I.K. Direct synthesis of hydrogen peroxide from hydrogen and oxygen over palladium catalyst supported on H3PW12O40-incorporated MCF silica. J. Mol. Catal. A Chem. 2011, 336, 78–86. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, T.J.; Chung, Y.M.; Oh, S.H.; Song, I.K. Direct synthesis of hydrogen peroxide from hydrogen and oxygen over insoluble Pd0.15M2.5H0.2PW12O40 (M = K, Rb, and Cs) heteropolyacid catalysts. Res. Chem. Intermed. 2010, 36, 639–646. [Google Scholar] [CrossRef]
- Park, S.Y.; Seo, J.G.; Jung, J.C.; Baeck, S.H.; Kim, T.J.; Chung, Y.M.; Oh, S.H.; Song, I.K. Direct synthesis of hydrogen peroxide from hydrogen and oxygen over palladium catalysts supported on TiO2–ZrO2 mixed metal oxides. Catal. Commun. 2009, 10, 1762–1765. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, T.J.; Chung, Y.M.; Oh, S.H.; Song, I.K. Direct Synthesis of Hydrogen Peroxide from Hydrogen and Oxygen over Palladium Catalyst Supported on SO3H-Functionalized SBA-15. Catal. Lett. 2009, 130, 296–300. [Google Scholar] [CrossRef]
- Park, S.Y.; Baeck, S.H.; Kim, T.J.; Chung, Y.M.; Oh, S.H.; Song, I.K. Direct synthesis of hydrogen peroxide from hydrogen and oxygen over palladium catalyst supported on SO3H-functionalized mesoporous silica. J. Mol. Catal. A Chem. 2010, 319, 98–107. [Google Scholar] [CrossRef]
- Blanco-Brieva, G.; Escrig, M.P.F.; Campos-Martin, J.M.G.; Fierro, J.L. Direct synthesis of hydrogen peroxide on palladium catalyst supported on sulfonic acid-functionalized silica. Green Chem. 2010, 12, 1163–1166. [Google Scholar] [CrossRef]
- Chung, Y.M.; Kwon, Y.T.; Kim, T.J.; Oh, S.H.; Lee, C.S. Direct synthesis of H2O2 catalyzed by Pd nanoparticles encapsulated in the multi-layered polyelectrolyte nanoreactors on a charged sphere. Chem. Commun. 2011, 47, 5705–5707. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Chung, Y.M.; Kang, S.M.; Choi, C.H.; Kim, B.Y.; Kwon, Y.T.; Kim, T.J.; Oh, S.H.; Lee, C.S. Palladium nanocatalysts immobilized on functionalized resin for the direct synthesis of hydrogen peroxide from hydrogen and oxygen. ACS Catal. 2012, 2, 1042–1048. [Google Scholar] [CrossRef]
- Blanco-Brieva, G.; Cano-Serrano, E.; Campos-Martin, J.M.; Fierro, J.L.G. Direct synthesis of hydrogen peroxide solution with palladium-loaded sulfonic acid polystyrene resins. Chem. Commun. 2004, 1184–1185. [Google Scholar] [CrossRef]
- Ntainjua, N.E.; Piccinini, M.; Pritchard, J.C.; Edwards, J.K.; Carley, A.F.; Moulijn, J.A.; Hutchings, G.J. Effect of halide and acid additives on the direct synthesis of hydrogen peroxide using supported Gold–Palladium catalysts. ChemSusChem 2009, 2, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.K.; Ntainjua, E.N.; Carley, A.F.; Herzing, A.A.; Kiely, C.J.; Hutchings, G.J. Direct synthesis of H2O2 from H2 and O2 over gold, palladium, and gold–palladium catalysts supported on acid-pretreated TiO2. Angew. Chem. Int. Ed. 2009, 48, 8512–8515. [Google Scholar] [CrossRef] [PubMed]
- Menegazzo, F.; Manzoli, M.; Signoretto, M.; Pinna, F.; Strukul, G. H2O2 direct synthesis under mild conditions on Pd-Au samples: Effect of the morphology and of the composition of the metallic phase. Catal. Today 2015, 248, 18–27. [Google Scholar] [CrossRef]
- Hiramatsu, Y.; Ishiuchi, Y.; Nagashima, H. Method for Producing Hydrogen Peroxide. U.S. Patent 5180573, 4 March 1993. [Google Scholar]
- Zhou, B.; Lee, L.-K. Catalyst and Process for Direct Catalytic Production of Hydrogen Peroxide. U.S. Patent 6,168,775, 2 January 2001. [Google Scholar]
- Gemo, N.; Biasi, P.; Canu, P.; Menegazzo, F.; Pinna, F.; Samikannu, A.; Kordás, K.; Salmi, T.; Mikkola, J.-P. Reactivity Aspects of SBA15-Based Doped Supported Catalysts: H2O2 Direct Synthesis and Disproportionation Reactions. Top. Catal. 2013, 56, 540–549. [Google Scholar] [CrossRef]
- Sun, M.; Zhang, J.Z.; Zhang, Q.H.; Wang, Y.; Wan, H.L. Polyoxometalate-supported Pd nanoparticles as efficient catalysts for the direct synthesis of hydrogen peroxide in the absence of acid or halide promoters. Chem. Commun. 2009, 5174–5176. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Lee, S.H.; Song, S.H.; Park, D.R.; Baeck, S.H.; Kim, T.J.; Chung, Y.M.; Oh, S.H.; Song, I.K. Direct synthesis of hydrogen peroxide from hydrogen and oxygen over palladium-exchanged insoluble heteropolyacid catalysts. Catal. Commun. 2009, 10, 391–394. [Google Scholar] [CrossRef]
- Desmedt, F.; Vlasselaer, Y.; Miquel, P. Direct Synthesis of Hydrogen Peroxide. WO Patent 072169, 15 May 2014. [Google Scholar]
- Desmedt, F.; Ganhy, J.P.; Vlasselaer, Y.; Miquel, P. A Catalyst for Direct Synthesis of Hydrogen Peroxide (Pd Reduced on Nb/Ta Oxide or Phosphate on Silica). WO Patent 068340, 16 May 2013. [Google Scholar]
- Fu, F.; Chuang, K.T.; Fiedorow, R. Selective oxidation of hydrogen to hydrogen peroxide. Stud. Surf. Sci. Catal. 1992, 72, 33–41. [Google Scholar] [CrossRef]
- Chuang, K.T.; Zhou, B. Production of Hydrogen Peroxide. U.S. Patent 5,338,531, 16 August 1994. [Google Scholar]
- Chuang, K.T. Production of Hydrogen Peroxide. U.S. Patent 5,082,647, 21 January 1992. [Google Scholar]
- Stakheev, A.Y.; Kustov, L.M. Effects of the support on the morphology and electronic properties of supported metal clusters: Modern concepts and progress in 1990s. Appl. Catal. A 1999, 188, 3–35. [Google Scholar] [CrossRef]
- Boyanov, B.I.; Morisson, T.I. Support and Temperature Effects in Platinum Clusters. 2. Electronic Properties. J. Phys. Chem. 1996, 100, 16318–16326. [Google Scholar] [CrossRef]
- Ramaker, D.E.; de Graaf, J.; Van Veen, J.A.R.; Koningsberger, D.C. Nature of the metal–support interaction in supported Pt catalysts: Shift in Pt valence orbital energy and charge rearrangement. J. Catal. 2001, 203, 7–17. [Google Scholar] [CrossRef]
- Stakheev, A.Y.; Shpiro, E.S.; Jaeger, N.I.; Schulz-Ekloff, G. Electronic state and location of Pt metal clusters in KL zeolite: FTIR study of CO chemisorption. Catal. Lett. 1995, 32, 147–158. [Google Scholar] [CrossRef]
- Ishihara, T.; Harada, K.; Eguchi, K.; Arai, H. Electronic interaction between supports and ruthenium catalysts for the hydrogenation of carbon monoxide. J. Catal. 1992, 136, 161–169. [Google Scholar] [CrossRef]
- Karpinski, Z.; Gandhi, S.N.; Sachtler, W.M.H. Neopentane Conversion Catalyzed by Pd in L-Zeolite: Effects of Protons, Ions, and Zeolite Structure. J. Catal. 1993, 141, 337–346. [Google Scholar] [CrossRef]
- Zhang, Z.; Wong, T.T.; Sachtler, W.M.H. The effect of Ca2+ and Mg2+ ions on the formation of electron-deficient palladium-proton adducts in zeolite Y. J. Catal. 1991, 128, 13. [Google Scholar] [CrossRef]
- Abate, S.; Centi, G.; Melada, S.; Perathoner, S.; Pinna, F.; Strukul, G. Preparation, performances and reaction mechanism for the synthesis of H2O2 from H2 and O2 based on palladium membranes. Catal. Today 2005, 104, 323–328. [Google Scholar] [CrossRef]
- Choudhary, V.R.; Samanta, C.; Gaikwad, A.G. Drastic increase of selectivity for H2O2 formation in direct oxidation of H2 to H2O2 over supported Pd catalysts due to their bromination. Chem. Commun. 2004, 2054–2055. [Google Scholar] [CrossRef] [PubMed]
- Samanta, C.; Choudhary, V.R. Direct synthesis of H2O2 from H2 and O2 over Pd/H-beta catalyst in an aqueous acidic medium: Influence of halide ions present in the catalyst or reaction medium on H2O2 formation. Catal. Commun. 2007, 8, 73–79. [Google Scholar] [CrossRef]
- Samanta, C.; Choudhary, V.R. Direct oxidation of H2 to H2O2 over Pd/Ga2O3 catalyst under ambient conditions: Influence of halide ions added to the catalyst or reaction medium. Appl. Catal. A 2007, 326, 28–36. [Google Scholar] [CrossRef]
- Samanta, C.; Choudhary, V.R. Direct formation of H2O2 from H2 and O2 and decomposition/hydrogenation of H2O2 in aqueous acidic reaction medium over halide-containing Pd/SiO2 catalytic system. Catal. Commun. 2007, 8, 2222–2228. [Google Scholar] [CrossRef]
- Samanta, C.; Choudhary, V.R. Direct synthesis of H2O2 from H2 and O2 and decomposition/hydrogenation of H2O2 in an aqueous acidic medium over halide-modified Pd/Al2O3 catalysts. Appl. Catal. A 2007, 330, 23–32. [Google Scholar] [CrossRef]
- Choudhary, V.R.; Samanta, C.; Jana, P. Method for Production of Hydrogen Peroxide with Improved Yield and Selectivity by Direct Oxidation of Hydrogen over Palladium Containing Catalyst. U.S. Patent 7,288,240, 30 October 2007. [Google Scholar]
- Jeong, H.E.; Kim, S.; Seo, M.G.; Lee, D.W.; Lee, K.Y. Catalytic activity of Pd octahedrons/SiO2 for the direct synthesis of hydrogen peroxide from hydrogen and oxygen. J. Mol. Catal. A Chem. 2016, 420, 88–95. [Google Scholar] [CrossRef]
- Choudhary, V.R.; Gaikwad, A.G. Kinetics of hydrogen peroxide decomposition in aqueous sulfuric acid over palladium/carbon: Effect of acid concentration. React. Kinet. Catal. Lett. 2003, 80, 27–32. [Google Scholar] [CrossRef]
- Wilson, N.M.; Flaherty, D.W. Mechanism for the Direct Synthesis of H2O2 on Pd Clusters: Heterolytic Reaction Pathways at the Liquid–Solid Interface. J. Am. Chem. Soc. 2016, 138, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, V.R.; Samanta, C. Role of chloride or bromide anions and protons for promoting the selective oxidation of H2 by O2 to H2O2 over supported Pd catalysts in an aqueous medium. J. Catal. 2006, 238, 28–38. [Google Scholar] [CrossRef]
- Samanta, C. Direct synthesis of hydrogen peroxide from hydrogen and oxygen: An overview of recent developments in the process. Appl. Catal. A 2008, 350, 133–149. [Google Scholar] [CrossRef]
- Abate, S.; Arrigo, R.; Schuster, M.E.; Perathoner, S.; Centi, G.; Villa, A.; Su, D.; Schlögl, R. Pd nanoparticles supported on N-doped nanocarbon for the direct synthesis of H2O2 from H2 and O2. Catal. Today 2010, 157, 280–285. [Google Scholar] [CrossRef]
- Abate, S.; Freni, M.; Arrigo, R.; Schuster, M.E.; Perathoner, S.; Centi, G. On the Nature of Selective Palladium-Based Nanoparticles on Nitrogen-Doped Carbon Nanotubes for the Direct Synthesis of H2O2. ChemCatChem 2013, 5, 1899–1905. [Google Scholar] [CrossRef]
- Arrigo, R.; Schuster, M.E.; Abate, S.; Wrabetz, S.; Amakawa, K.; Teschner, D.; Freni, M.; Centi, G.; Perathoner, S.; Hävecker, M.; et al. Dynamics of Palladium on Nanocarbon in the Direct Synthesis of H2O2. ChemSusChem 2014, 7, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, M.; Ikezawa, A.; Tobimatsu, H.; Irie, H.; Hashimoto, K. Zeta potential and photocatalytic activity of nitrogen doped TiO2 thin films. Phys. Chem. Chem. Phys. 2004, 6, 865–870. [Google Scholar] [CrossRef]
- Li, Q.; Easter, N.; Shang, J.K. As(III) Removal by Palladium-Modified Nitrogen-Doped Titanium Oxide Nanoparticle Photocatalyst. Environ. Sci. Technol. 2009, 43, 1534–1539. [Google Scholar] [CrossRef] [PubMed]
- Gemo, N.; Menegazzo, F.; Biasi, P.; Sarkar, A.; Samikannu, A.; Raut, D.G.; Kordas, K.; Rautio, A.; Mohl, M.; Bostrom, D.; et al. TiO2 nanoparticles vs. TiO2 nanowires as support in hydrogen peroxide direct synthesis: The influence of N and Au doping. RSC Adv. 2016, 6, 103311–103319. [Google Scholar] [CrossRef]
- Freakley, S.J.; Lewis, R.J.; Morgan, D.J.; Edwards, J.K.; Hutchings, G.J. Direct synthesis of hydrogen peroxide using Au–Pd supported and ion-exchanged heteropolyacids precipitated with various metal ions. Catal. Today 2015, 248, 10–17. [Google Scholar] [CrossRef]
- Gudarzi, D.; Ratchananusorn, W.; Turunen, I.; Heinonen, M.; Salmi, T. Factors affecting catalytic destruction of H2O2 by hydrogenation and decomposition over Pd catalysts supported on activated carbon cloth (ACC). Catal. Today 2015, 248, 69–79. [Google Scholar] [CrossRef]
- Dissanayake, D.P.; Lunsford, J.H. The direct formation of H2O2 from H2 and O2 over colloidal palladium. J. Catal. 2003, 214, 113–120. [Google Scholar] [CrossRef]
- Seo, M.; Lee, D.; Han, S.S.; Lee, K. Direct synthesis of hydrogen peroxide from hydrogen and oxygen over mesoporous silica-shell-coated, palladium-nanocrystal-grafted SiO2 Nanobeads. ACS Catal. 2017, 7, 3039–3048. [Google Scholar] [CrossRef]
- Arrigo, R.; Schuster, M.E.; Abate, S.; Giorgianni, G.; Centi, G.; Perathoner, S.; Wrabetz, S.; Pfeifer, V.; Antonietti, M.; Schlog, R. Pd Supported on Carbon Nitride Boosts the Direct Hydrogen Peroxide Synthesis. ACS Catal. 2016, 6, 6959–6966. [Google Scholar] [CrossRef]
- Abate, S.; Perathoner, S.; Genovese, C.; Centi, G. Performances, characteristics and stability of catalytic membranes based on a thin Pd film on a ceramic support for H2O2 direct synthesis. Desalination 2006, 200, 760–761. [Google Scholar] [CrossRef]
- Pashkova, A.; Svajda, K.; Dittmeyer, R. Direct synthesis of hydrogen peroxide in a catalytic membrane contactor. Chem. Eng. Sci. 2008, 139, 165–171. [Google Scholar] [CrossRef]
- Wang, L.; Bao, S.G.; Yi, J.H.; He, F.; Mi, Z. Preparation and properties of Pd/Ag composite membrane for direct synthesis of hydrogen peroxide from hydrogen and oxygen. Appl. Catal. B 2008, 79, 157–162. [Google Scholar] [CrossRef]
- Inoue, T.; Tanaka, Y.; Tanaka, D.A.P.; Suzuki, T.M.; Sato, K.; Nishioka, M.; Hamakawa, S.; Mizukami, F. Direct production of hydrogen peroxide from oxygen and hydrogen applying membrane-permeation mechanism. Chem. Eng. Sci. 2010, 65, 436–440. [Google Scholar] [CrossRef]
- Shi, L.; Goldbach, A.; Zeng, G.F.; Xu, H.Y. Direct H2O2 synthesis over Pd membranes at elevated temperatures. J. Membr. Sci. 2010, 348, 160–166. [Google Scholar] [CrossRef]
- Shi, L.; Goldbach, A.; Zeng, G.F.; Xu, H.Y. H2O2 synthesis over PdAu membranes. Catal. Today 2010, 156, 118–123. [Google Scholar] [CrossRef]
- Pashkova, A.; Dittmeyer, R.; Kaltenborn, N.; Richter, H. Experimental study of porous tubular catalytic membranes for direct synthesis of hydrogen peroxide. Chem. Eng. J. 2010, 165, 924–933. [Google Scholar] [CrossRef]
- Tian, P.; Ouyang, L.; Xu, X.; Xu, J.; Han, Y.F. Density functional theory study of direct synthesis of H2O2 from H2 and O2 on Pd(111), Pd(100), and Pd(110) surfaces. Chin. J. Catal. 2013, 34, 1002–1012. [Google Scholar] [CrossRef]
- Kim, S.; Lee, D.W.; Lee, K.Y. Shape-dependent catalytic activity of palladium nanoparticles for the direct synthesis of hydrogen peroxide from hydrogen and oxygen. J. Mol. Catal. A Chem. 2014, 391, 48–54. [Google Scholar] [CrossRef]
- Kim, S.; Lee, D.W.; Lee, K.Y. Direct synthesis of hydrogen peroxide from hydrogen and oxygen over single-crystal cubic palladium on silica catalysts. J. Mol. Catal. A Chem. 2014, 383–384, 64–69. [Google Scholar] [CrossRef]
- Kim, S.; Lee, D.W.; Lee, K.Y.; Cho, E. Effect of Pd Particle Size on the Direct Synthesis of Hydrogen Peroxide from Hydrogen and Oxygen over Pd Core–Porous SiO2 Shell Catalysts. Catal. Lett. 2014, 144, 905–911. [Google Scholar] [CrossRef]
- Seo, M.G.; Kim, S.; Lee, D.W.; Jeong, H.E.; Lee, K.Y. Core–shell structured, nano-Pd-embedded SiO2–Al2O3 catalyst (Pd@SiO2–Al2O3) for direct hydrogen peroxide synthesis from hydrogen and oxygen. Appl. Catal. A 2016, 511, 87–94. [Google Scholar] [CrossRef]
- Lee, H.; Kim, S.; Lee, D.W.; Lee, K.Y. Direct synthesis of hydrogen peroxide from hydrogen and oxygen over a Pd core-silica shell catalyst. Catal. Commun. 2011, 12, 968–971. [Google Scholar] [CrossRef]
- Joo, S.H.; Park, J.Y.; Tsung, C.K.; Yamada, Y.; Yang, P.; Somorjai, G.A. Thermally stable Pt/mesoporous silica core–shell nanocatalysts for high-temperature reactions. Nat. Mater. 2009, 8, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.H.; Doong, R.A. Bifunctional Au−Fe3O4 Heterostructures for Magnetically Recyclable Catalysis of Nitrophenol Reduction. J. Phys. Chem. C 2011, 115, 6591–6598. [Google Scholar] [CrossRef]
- Qi, J.; Chen, J.; Li, G.; Li, S.; Gao, Y.; Tang, Z. Facile synthesis of core–shell Au@CeO2 nanocomposites with remarkably enhanced catalytic activity for CO oxidation. Energy Environ. Sci. 2012, 5, 8937–8941. [Google Scholar] [CrossRef]
- Yin, H.; Ma, Z.; Chi, M.; Dai, S. Heterostructured catalysts prepared by dispersing Au@Fe2O3 core–shell structures on supports and their performance in CO oxidation. Catal. Today 2011, 160, 87–95. [Google Scholar] [CrossRef]
- Tripathy, S.K.; Mishra, A.; Jha, S.K.; Wahab, R.; Al-Khedhairy, A.A. Synthesis of thermally stable monodispersed Au@SnO2 core–shell structure nanoparticles by a sonochemical technique for detection and degradation of acetaldehyde. Anal. Meth. 2013, 5, 1456–1462. [Google Scholar] [CrossRef]
- Meir, N.; Jen-La Plante, I.; Flomin, K.; Chockler, E.; Moshofsky, B.; Diab, M.; Volokh, M.; Mokari, T. Studying the chemical, optical and catalytic properties of noble metal (Pt, Pd, Ag, Au)–Cu2O core–shell nanostructures grown via a general approach. J. Mater. Chem. A 2013, 1, 1763–1769. [Google Scholar] [CrossRef]
- Han, L.; Zhu, C.; Hu, P.; Dong, S. One-pot synthesis of a Au@TiO2 core–shell nanocomposite and its catalytic property. RSC Adv. 2013, 3, 12568–12570. [Google Scholar] [CrossRef]
- Liu, W.; Zhong, W.; Jiang, H.Y.; Tang, N.J.; Wu, X.L.; Du, W.Y. Synthesis and magnetic properties of FeNi3/Al2O3 core-shell nanocomposites. Eur. Phys. J. B 2005, 46, 471–474. [Google Scholar] [CrossRef]
- Sun, H.; He, J.; Wang, J.; Zhang, S.Y.; Liu, C.; Sritharan, T.; Mhaisalkar, S.; Han, M.Y.; Wang, D.; Chen, H. Investigating the Multiple Roles of Polyvinylpyrrolidone for a General Methodology of Oxide Encapsulation. J. Am. Chem. Soc. 2013, 135, 9099–9110. [Google Scholar] [CrossRef] [PubMed]
- Ntainjua, N.E.; Edwards, J.K.; Carley, A.F.; Lopez-Sanchez, J.A.; Moulijn, J.A.; Herzing, A.A.; Kiely, C.J.; Hutchings, G.J. The role of the support in achieving high selectivity in the direct formation of hydrogen peroxide. Green Chem. 2008, 10, 1162–1169. [Google Scholar] [CrossRef]
- Seo, M.; Kim, H.J.; Han, S.S.; Lee, K. Effect of shell thickness of Pd core-porous SiO2 shell catalysts on direct synthesis of H2O2 from H2 and O2. J. Mol. Catal. A Chem. 2017, 426, 238–243. [Google Scholar] [CrossRef]
- Sterchele, S.; Biasi, P.; Centomo, P.; Campestrini, S.; Shchukarev, A.; Rautio, A.; Mikkola, J.; Salmi, T.; Zecca, M. The effect of the metal precursor-reduction with hydrogen on a library of bimetallic Pd-Au and Pd-Pt catalysts for the direct synthesis of H2O2. Catal. Today 2015, 248, 40–47. [Google Scholar] [CrossRef]
- Pritchard, J.; Piccinini, M.; Tiruvalam, R.; He, Q.; Dimitratos, N.; Lopez-Sanchez, J.A.; Morgan, D.J.; Carley, A.F.; Edwards, J.K.; Kiely, C.J.; et al. Effect of heat treatment on Au-Pd catalysts synthesized by sol immobilisation for the direct synthesis of hydrogen peroxide and benzyl alcohol oxidation. Catal. Sci. Technol. 2013, 3, 308–317. [Google Scholar] [CrossRef]
- Pritchard, J.; Kesavan, L.; Piccinini, M.; He, Q.; Tiruvalam, R.; Dimitratos, N.; Lopez-Sanchez, J.A.; Carley, A.F.; Edwards, J.K.; Kiely, C.J.; et al. Direct Synthesis of Hydrogen Peroxide and Benzyl Alcohol Oxidation Using Au-Pd Catalysts Prepared by Sol Immobilization. Langmuir 2010, 26, 16568–16577. [Google Scholar] [CrossRef] [PubMed]
- Tiruvalam, R.C.; Pritchard, J.C.; Dimitratos, N.; Lopez-Sanchez, J.A.; Edwards, J.K.; Carley, A.F.; Hutchings, G.J.; Kiely, C.J. Aberration corrected analytical electron microscopy studies of sol-immobilized Au + Pd, Au{Pd} and Pd{Au} catalysts used for benzyl alcohol oxidation and hydrogen peroxide production. Faraday Discuss. 2011, 152, 63–86. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.G.; Kim, S.; Jeong, H.E.; Lee, D.W.; Lee, K.Y. A yolk–shell structured Pd@void@ZrO2 catalyst for direct synthesis of hydrogen peroxide from hydrogen and oxygen. J. Mol. Catal. A Chem. 2016, 413, 1–6. [Google Scholar] [CrossRef]
- Chen, Z.; Pan, H.; Lin, Q.; Zhang, X.; Xiao, S.; He, S. The modification of Pd core–silica shell catalysts by functional molecules (KBr, CTAB, SC) and their application to the direct synthesis of hydrogen peroxide from hydrogen and oxygen. Catal. Sci. Technol. 2017, 7, 1415–1422. [Google Scholar] [CrossRef]
- Lari, G.M.; Puertolas, B.; Shahrokhi, M.; Lopez, N.; Perez-Ramirez, J. Hybrid Palladium Nanoparticles for Direct Hydrogen Peroxide Synthesis: The Key Role of the Ligand. Angew. Chem. Int. Ed. 2017, 56, 1775–1779. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Jeong, H.; Chung, Y. Direct synthesis of hydrogen peroxide over Pd/C catalyst prepared by selective adsorption deposition method. J. Catal. 2018, 365, 125–137. [Google Scholar] [CrossRef]
- Hu, B.Z.; Deng, W.P.; Li, R.S.; Zhang, Q.; Wang, Y.; Delplanque-Janssens, F.; Paul, D.; Desmedt, F.; Mique, P. Carbon-supported palladium catalysts for the direct synthesis of hydrogen peroxide from hydrogen and oxygen. J. Catal. 2014, 319, 15–26. [Google Scholar] [CrossRef]
- Mori, K.; Araki, T.; Shironita, S.; Sonoda, J.; Yamashita, H. Supported Pd and PdAu Nanoparticles on Ti-MCM-41 Prepared by a Photo-assisted Deposition Method as Efficient Catalysts for Direct Synthesis of H2O2 from H2 and O2. Catal. Lett. 2009, 131, 337–343. [Google Scholar] [CrossRef]
- Mori, K.; Furubayashi, K.; Okada, S.; Yamashita, H. Synthesis of Pd nanoparticles on heteropolyacid-supported silica by a photo-assisted deposition method: An active catalyst for the direct synthesis of hydrogen peroxide. RSC Adv. 2012, 2, 1047–1054. [Google Scholar] [CrossRef]
- Howe, A.; Miedziak, P.; Morgan, D.J.; He, Q.; Strasser, P.; Edwards, J. One pot microwave synthesis of highly stable AuPd@Pd supported core-shell nanoparticles. Faraday Discuss. 2018, 208, 409–425. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Seo, M.; Cho, Y.; Han, S.S.; Lee, K. Highly dispersed Pd catalysts prepared by a sonochemical method for the direct synthesis of hydrogen peroxide. J. Mol. Catal. A Chem. 2017, 429, 43–50. [Google Scholar] [CrossRef]
- Sierra-Salazar, A.F.; Li, W.S.J.; Bathfield, M.; Ayral, A.; Abate, S.; Chave, T.; Nikitenko, S.I.; Hulea, V.; Perathoner, S.; Lacroix-Desmazes, P. Hierarchically porous Pd/SiO2 catalyst by combination of miniemulsion polymerisation and sol-gel method for the direct synthesis of H2O2. Catal. Today 2018, 306, 16–22. [Google Scholar] [CrossRef]
- Gopal, R. Electrochemical Synthesis of Hydrogen Peroxide. U.S. Patent 0019758 A1, 22 January 2003. [Google Scholar]
- Yi, Y.; Zhou, J.; Guo, H.; Zhao, J.; Su, J.; Wang, L.; Wang, X.; Gong, W. Safe direct synthesis of high purity H2O2 through a H2/O2 plasma reaction. Angew. Chem. Int. Ed. 2013, 52, 8446–8449. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The different reactions involved when reacting hydrogen and oxygen.

Figure 2.
The convergence of the different features necessary to define the “dream catalyst”.

Figure 3.
A schematic representation of the different alloying metals/promoters used to improve Pd performance. Areas approximately correspond to their occurrence in the literature.
Figure 3.
A schematic representation of the different alloying metals/promoters used to improve Pd performance. Areas approximately correspond to their occurrence in the literature.

Figure 4.
Effect of the presence or absence of a second metal in O2 and H2O2 chemisorption leading respectively to hydrogen peroxide or water formation.
Figure 4.
Effect of the presence or absence of a second metal in O2 and H2O2 chemisorption leading respectively to hydrogen peroxide or water formation.

Figure 5.
Catalytic performance of Pd catalysts as a function of the preparation temperature (A) and the heteropolyacid content (B) according to Ref. 90. A: conversion (squares), yield (diamonds); B: selectivity (squares), yield (diamonds).
Figure 5.
Catalytic performance of Pd catalysts as a function of the preparation temperature (A) and the heteropolyacid content (B) according to Ref. 90. A: conversion (squares), yield (diamonds); B: selectivity (squares), yield (diamonds).

Figure 6.
The role of support acidity in stabilizing H2O2 and promoting H2O2 formation. Mesopores facilitate H2O2 back-diffusion decreasing the probability of decomposition.
Figure 6.
The role of support acidity in stabilizing H2O2 and promoting H2O2 formation. Mesopores facilitate H2O2 back-diffusion decreasing the probability of decomposition.

Figure 7.
The different morphologies of the catalysts reported in this paragraph.

Figure 8.
The process of core@shell structure formation.

Figure 9.
Roles of surface morphology and additives.

Figure 10.
A schematic reprentation of the synthetic protocol for the preparation of hierarchically porous Pd/SiO2 catalysts.
Figure 10.
A schematic reprentation of the synthetic protocol for the preparation of hierarchically porous Pd/SiO2 catalysts.


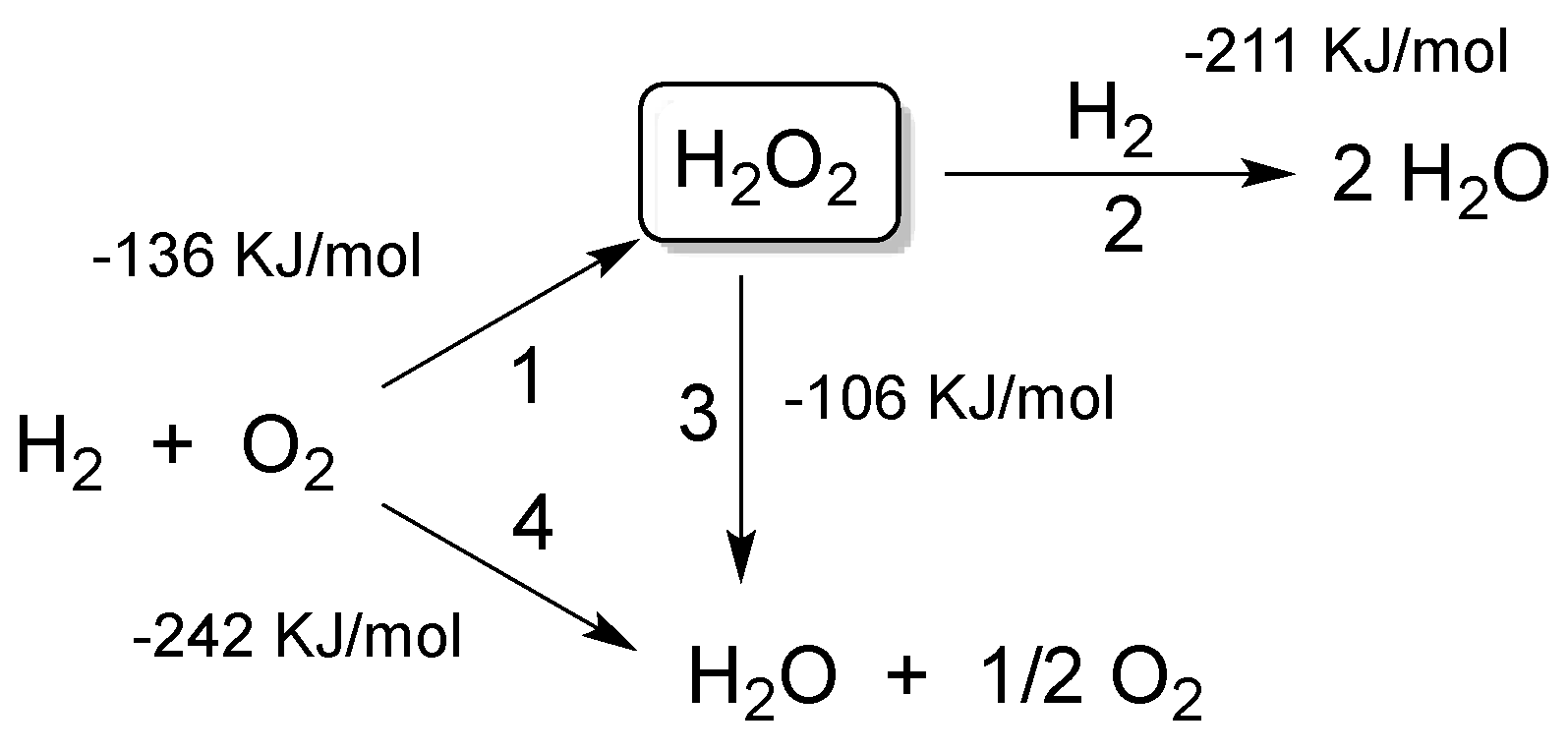{kind=link}
{kind=link}
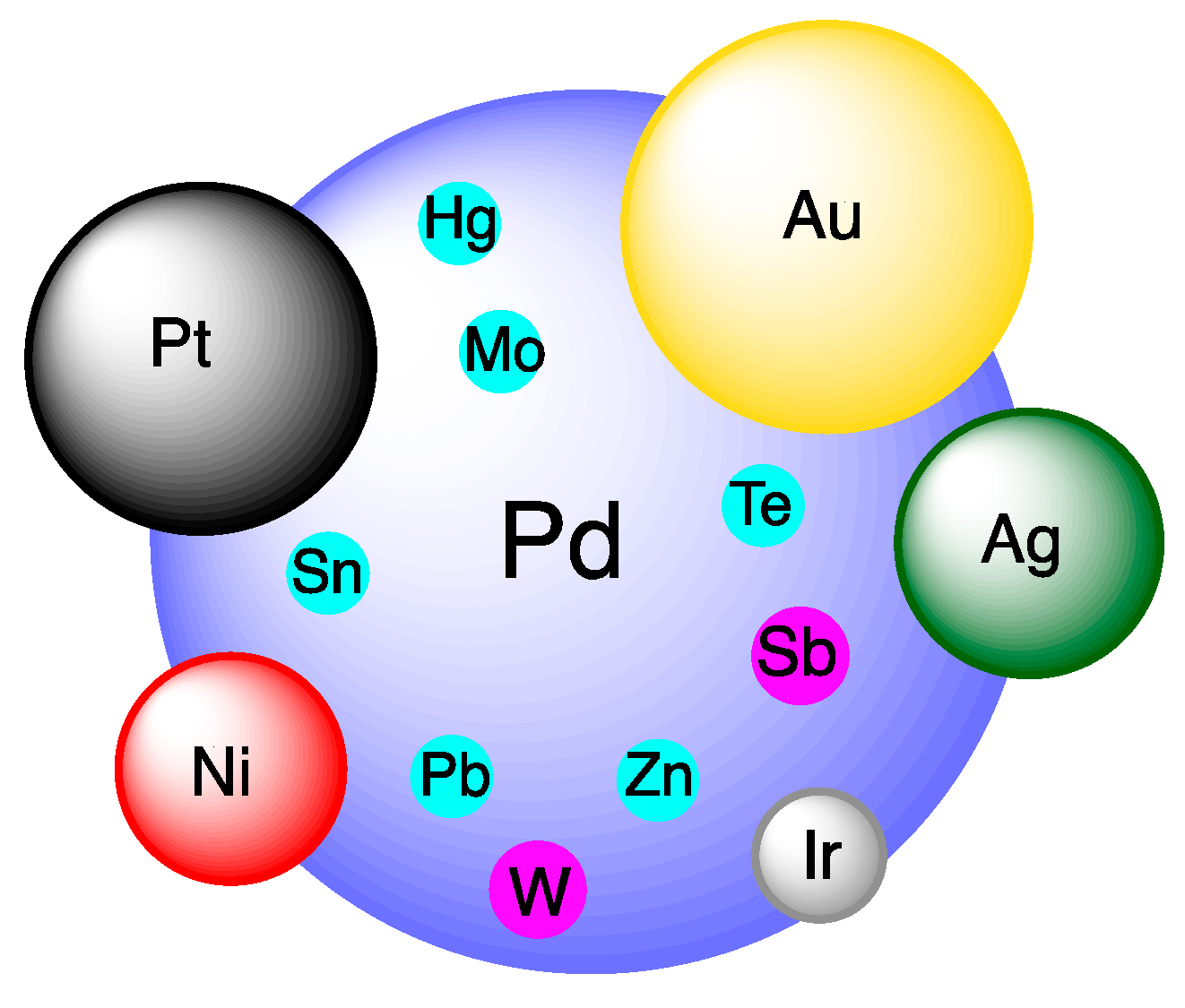{kind=link}
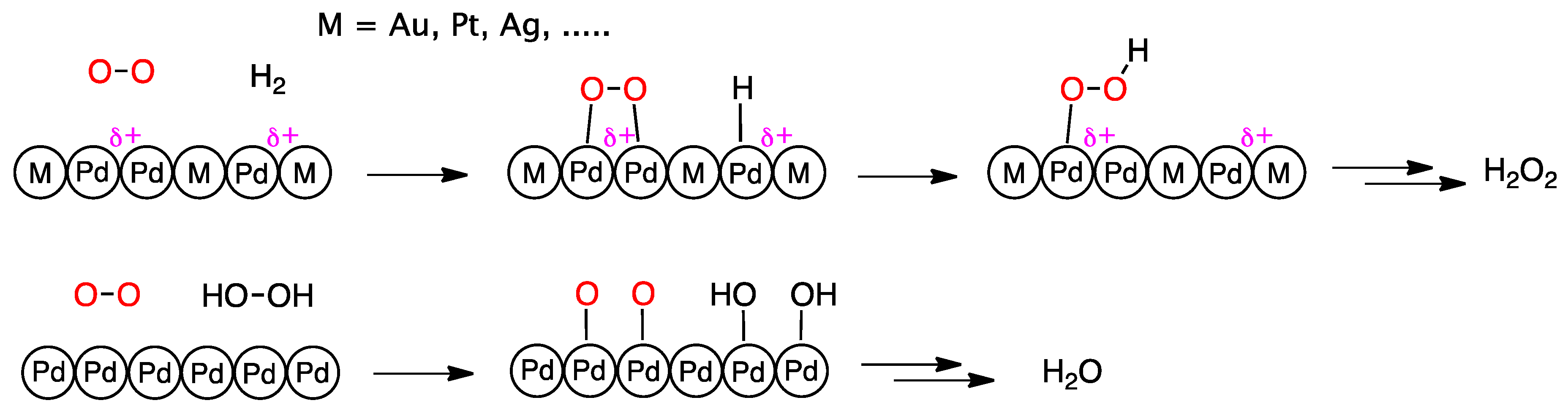{kind=link}
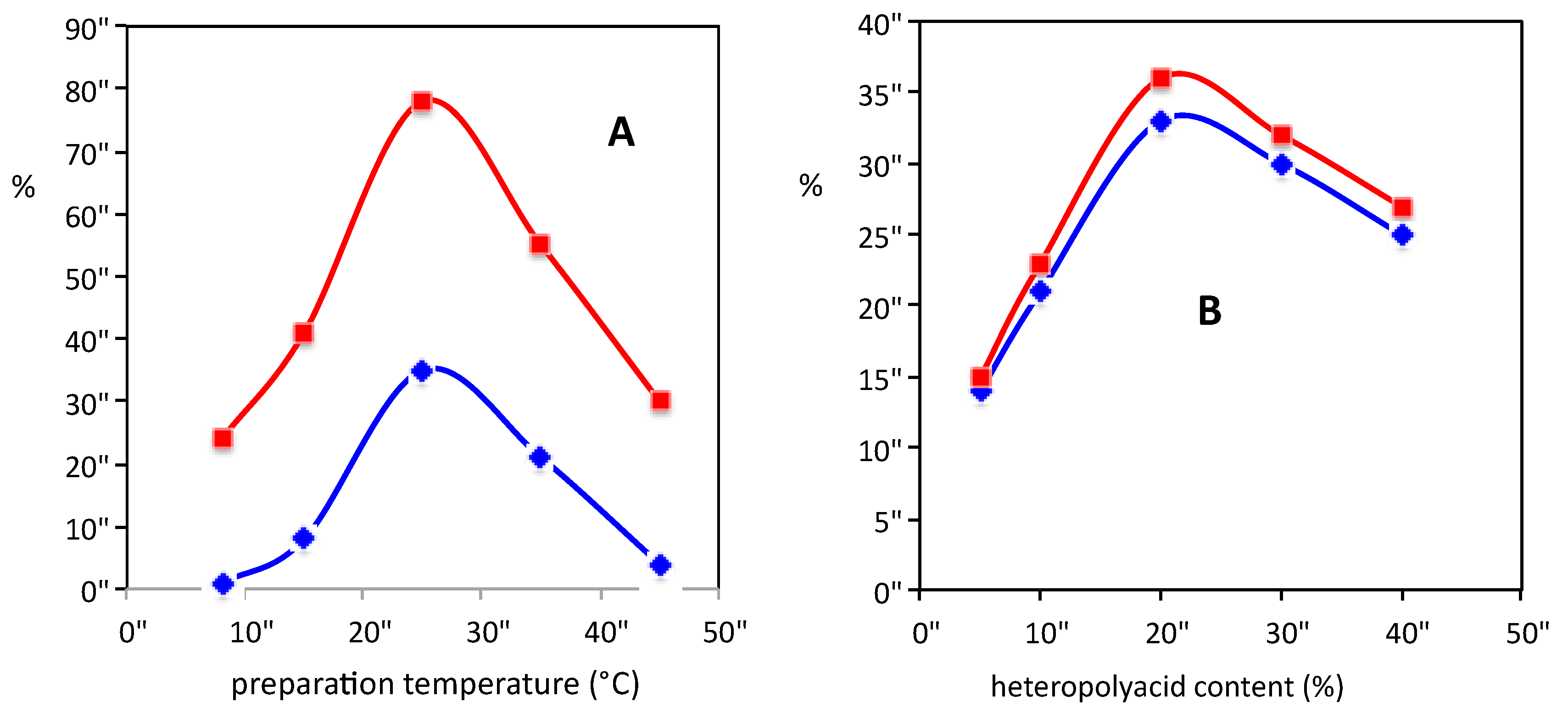{kind=link}
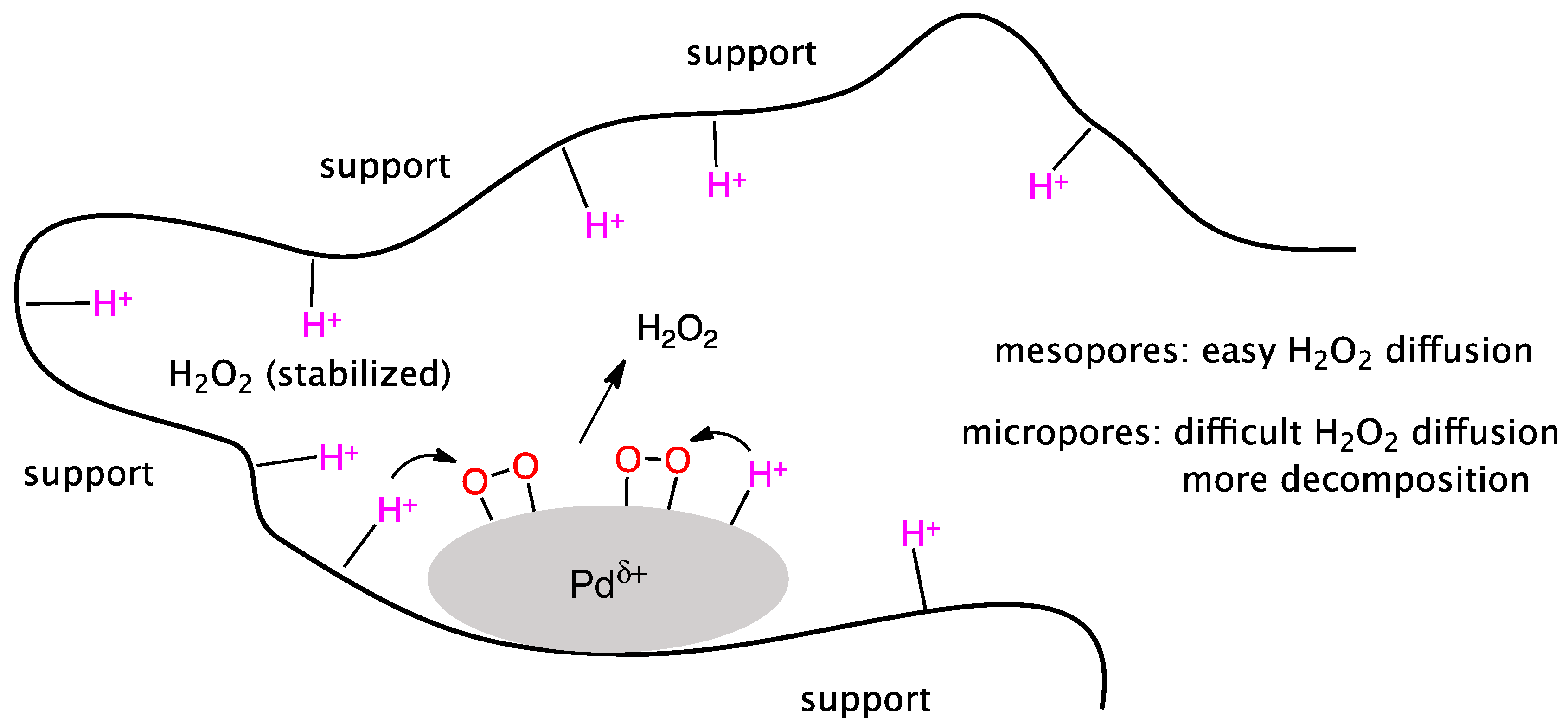{kind=link}
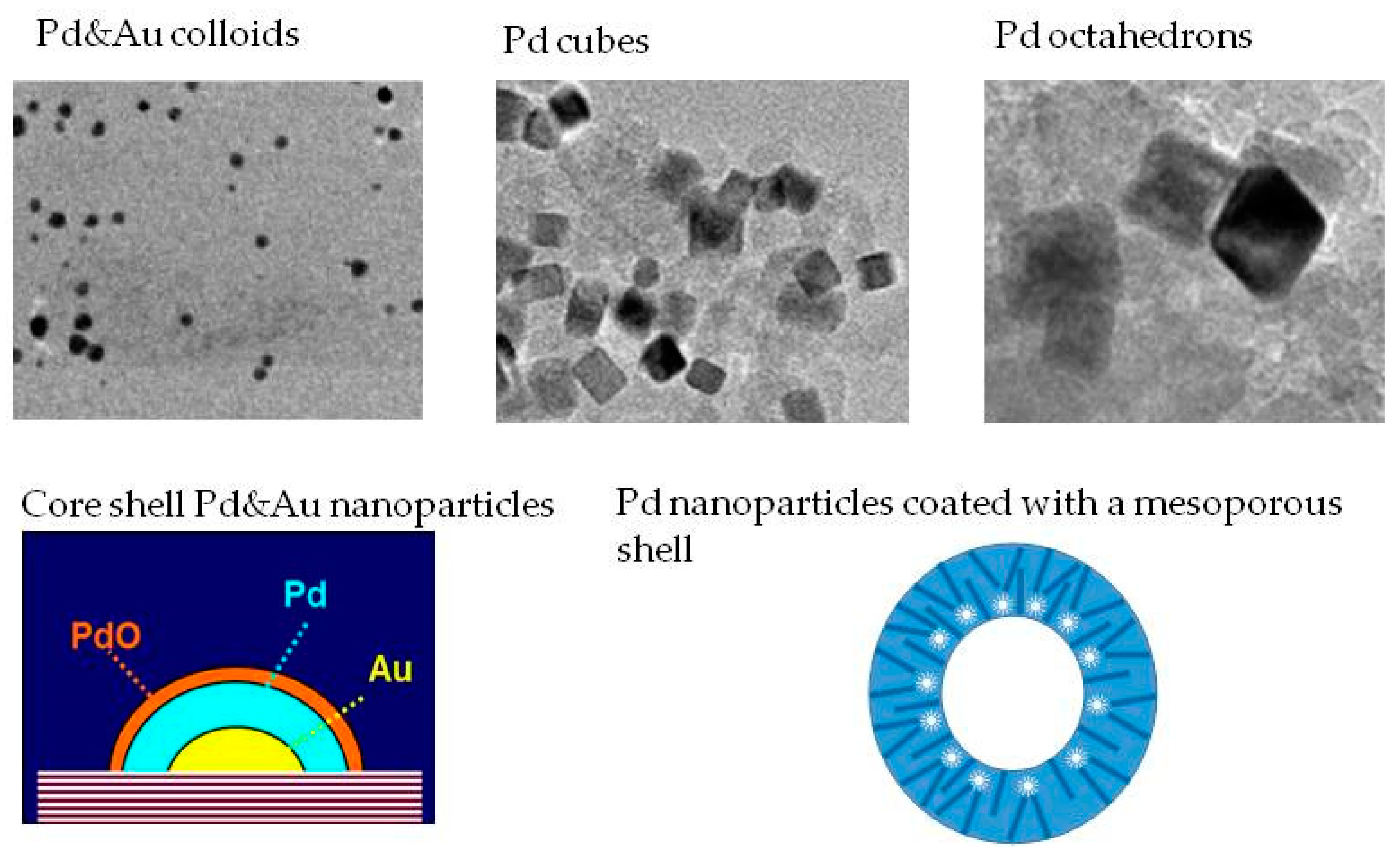{kind=link}
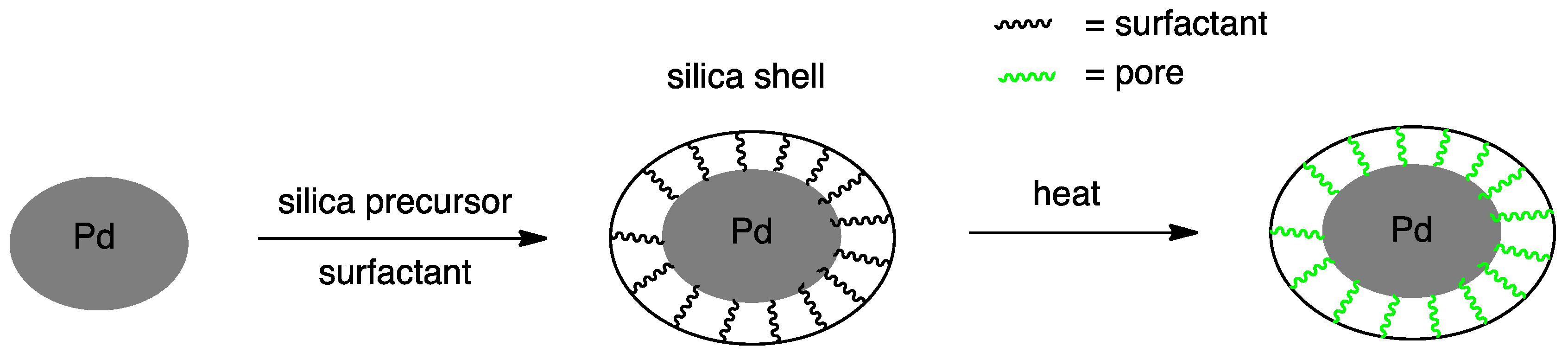{kind=link}
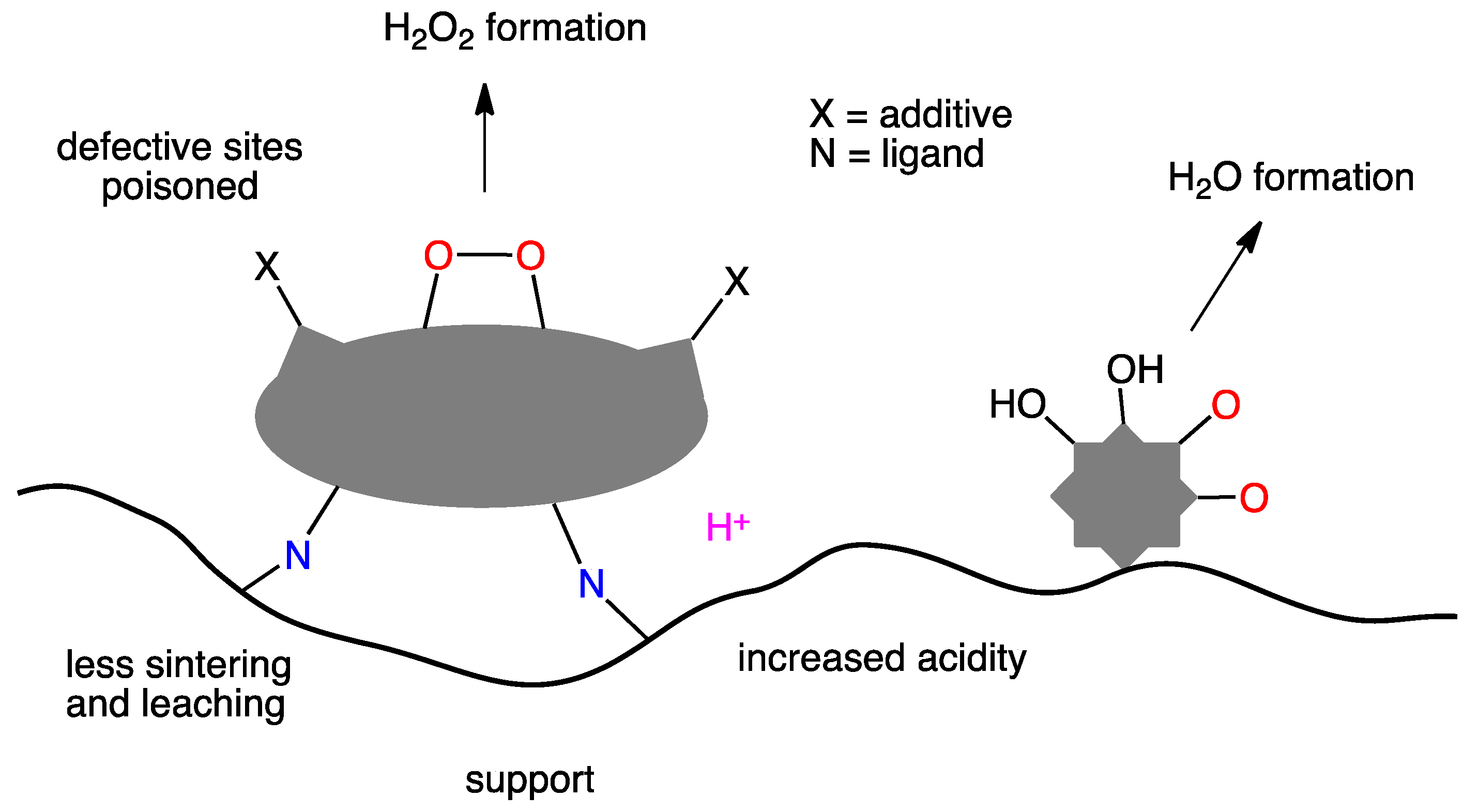{kind=link}
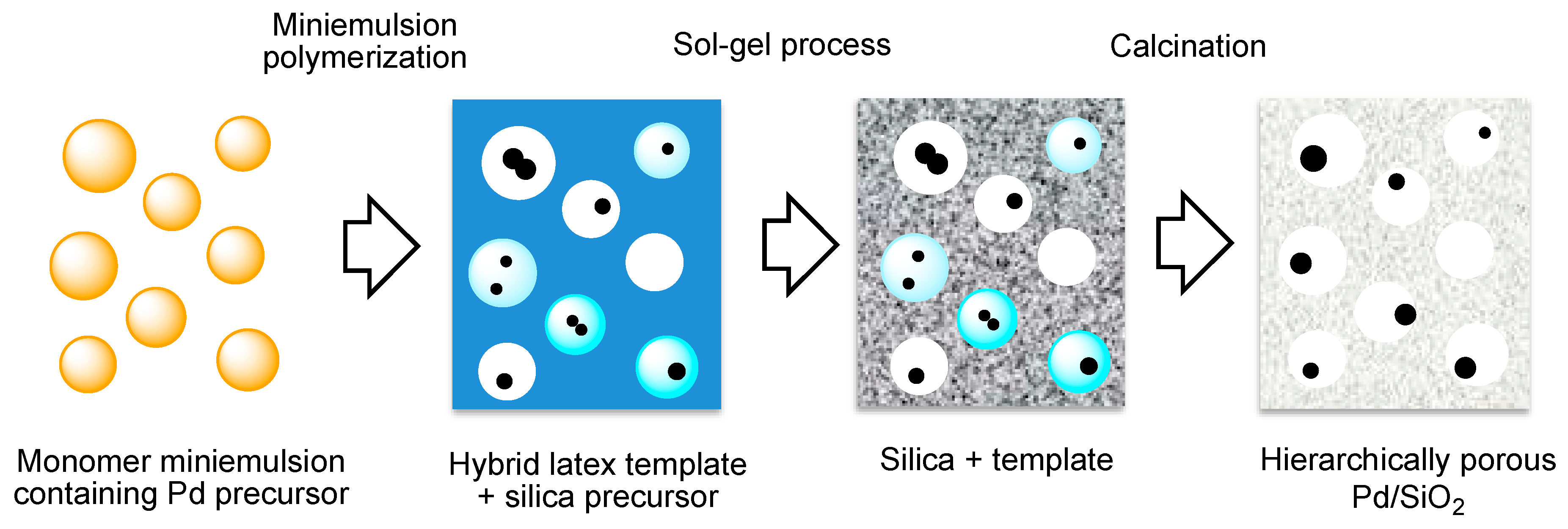{kind=link}
Table 1.
Some representative examples of metal promoted/alloyed catalysts and their use in the direct oxidation of hydrogen peroxide.
Table 1.
Some representative examples of metal promoted/alloyed catalysts and their use in the direct oxidation of hydrogen peroxide.
| Active Phase | Support | Solvent | Time (min) | Temp (°C) | P (bar) | Activity mol/KgMeh | Productivity mol/KgM | Sel (%) | Ref |
|---|---|---|---|---|---|---|---|---|---|
| 1% Pd + Ag Pd/Ag 40/1 | AC | MeOH/H2SO4 | 15 | 2 | 30 | 7022 | 1755 | 71 | 53 |
| 1.25%Pd + 1.25%Au | SZ | MeOH/H2SO4 | 180 | 20 | 1 | 635 | 1905 | 61 | 41 |
| 2.5%Pd + 2.5%Au | TiO2 | MeOH/H2O | 30 | 2 | 37 | 1280 | 640 | 60 | 54 |
| 2.5%Pd + 2.5%Au | AcC | MeOH/H2O | 30 | 2 | 37 | 3200 | 1600 | 95 | 57 |
| 5%Au | TiO2 | MeOH/H2O | 30 | 2 | 37 | 9.6 | 4.8 | 95 | |
| 3.3%Pd + 9%Au | SiO2 | EtOH/HCl | 300 | 10 | 1 | 510 | 2554 | 59 | 67 |
| 1%Pd + 2%Au | OAC | MeOH | 180 | 0 | 38 | 17,133 | 51,400 | 63 | 68 |
| 3.3%Pd + 1.7%Au | SiO2 | MeOH/H2SO4 | 60 | 25 | 20 | 570 | 570 | 35 | 69 |
| 3.3%Pd + 0.32%Pt | SiO2 | EtOH/HCl | 300 | 10 | 1 | 1496 | 7481 | 70 | 70 |
| 1.3%Pd + 0.2%Pt | SZ | MeOH/H2SO4 | 300 | 25 | 1 | 1074 | 5374 | 70 | 76 |
| 2.4%Pd + 2.4%Au + 0.2%Pt | CeO2 | MeOH/H2O | 30 | 2 | 40 | 3400 | 1700 | 79 | |
| 0.6%Pd + 0.4%Ni | // | H2O/HCl/Br− | 72 h | 10 | 1 | 30 | 2136 | 95 | 80 |
| 1%Pd + 5%Zn | γ-Al2O3 | MeOH/H2SO4 | 15 | 2 | 30 | 4238 | 1059 | 76 | 84 |
| 3%Pd + 0.03%Te | TiO2 | EtOH/H2SO4 | 60 | 5 | 1 | 977 | 977 | 100 | 87 |
Legend for supports: AC activated carbon; SZ sulfated zirconia; AcC acid pretreated carbon; OAC oxidized activated carbon.
Table 2.
The performance of some Pd and Pd/Au catalysts on different supports.
| Active Phase | Support | Solvent | Time (min) | Temp (°C) | P (bar) | Activity mol/KgMeh | Productivity mol/KgM | Sel (%) | Ref |
|---|---|---|---|---|---|---|---|---|---|
| 1.5% Pd | SiO2 | MeO/H2SO4 | 30 | 20 | 10 | 14,000 | 28,000 | 64 | 85 |
| 2.5t% Pd + 2.5% Au | carbon | MeOH/H2O | 30 | 2 | 37 | 2200 | 4400 | 95 | |
| 2.5t% Pd + 2.5% Au | A CMK-3 C | MeOH/HCl | 60 | 5 | 20 | 640 | 640 | 99 | 98 |
| 2.5t% Pd + 2.5% Au | MgO | MeOH/H3PO4 | 30 | 2 | 37 | 3980 | 7960 | 117 | |
| 2.5% Pd | SBA15 | MeOH/H2SO4 | 30 | 25 | 10 | 8000 | 16,000 | 60 | 101 |
| 3.2% Pd + 1.8% Au | SBA15-NH2-SO3H | MeOH/H2O | 240 | 2 | 40 | 750 | 3000 | 102 | |
| 0.3% Pd | ZrCeLaOx | MeOH | 300 | 10 | 50 | 3759 | 18,796 | 15 | 103 |
Legend for supports: A CMK-3 C acid pre-treated CMK-3 carbon; SBA15-NH2-SO3H is a SBA15 functionalized with both -SO3H and -NH2 organic groups; ZrCeLaOx is a mixed ZrO2/CeO2/La2O3 77.5/17.5/5 oxide.
Table 3.
The effect of the acidity of different supports on Pd catalysts.
| Active Phase | Support | Solvent | Time (min) | Temp (°C) | P (bar) | Activity mol/KgMh | Productivity mol/KgM | Sel (%) | Ref |
|---|---|---|---|---|---|---|---|---|---|
| 0.45% Pd | HZSM-5-30 | MeOH/NaBr | 360 | 28 | 10 | 5 | 31 | 15 | 104 |
| 0.97% Pd | SiO2-SO3H | MeOH/HBr | 120 | 35 | 95 | 11,340 | 22,680 | 65 | 113 |
| 1.49% Pd | PS-SO3H | MeOH/HBr | 120 | 35 | 100 | 10,385 | 20,770 | 77 | 116 |
| 1% Pd | Cs1.5H1.5PW12O40 | EtOH | 240 | 10 | 1 | 7650 | 30,600 | 67 | 123 |
Legend for supports: SiO2-SO3H sulfonic acid-functionalized silica; PS-SO3H sulfonated-polystyrene resins; Cs1.5H1.5PW12O40 phosphotungstate heteropolyacid cesium salt.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Menegazzo, F.; Signoretto, M.; Ghedini, E.; Strukul, G. Looking for the “Dream Catalyst” for Hydrogen Peroxide Production from Hydrogen and Oxygen. Catalysts 2019, 9, 251. https://doi.org/10.3390/catal9030251
AMA Style
Menegazzo F, Signoretto M, Ghedini E, Strukul G. Looking for the “Dream Catalyst” for Hydrogen Peroxide Production from Hydrogen and Oxygen. Catalysts. 2019; 9(3):251. https://doi.org/10.3390/catal9030251
Chicago/Turabian StyleMenegazzo, Federica, Michela Signoretto, Elena Ghedini, and Giorgio Strukul. 2019. "Looking for the “Dream Catalyst” for Hydrogen Peroxide Production from Hydrogen and Oxygen" Catalysts 9, no. 3: 251. https://doi.org/10.3390/catal9030251
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.