
Single-Crystal X-ray Structure Determination of Tris(pyrazol-1-yl)methane Triphenylphosphine Copper(I) Tetrafluoroborate, Hirshfeld Surface Analysis and DFT Calculations

Abstract
:1. Introduction
2. Experimental Section
2.1. Materials and Methods
2.2. Synthesis of [Cu(CHpz3)(PPh3)][BF4]
2.3. Crystal Structure Determination and Hirshfeld Surface Analysis
2.4. Computational Details
3. Results and Discussion
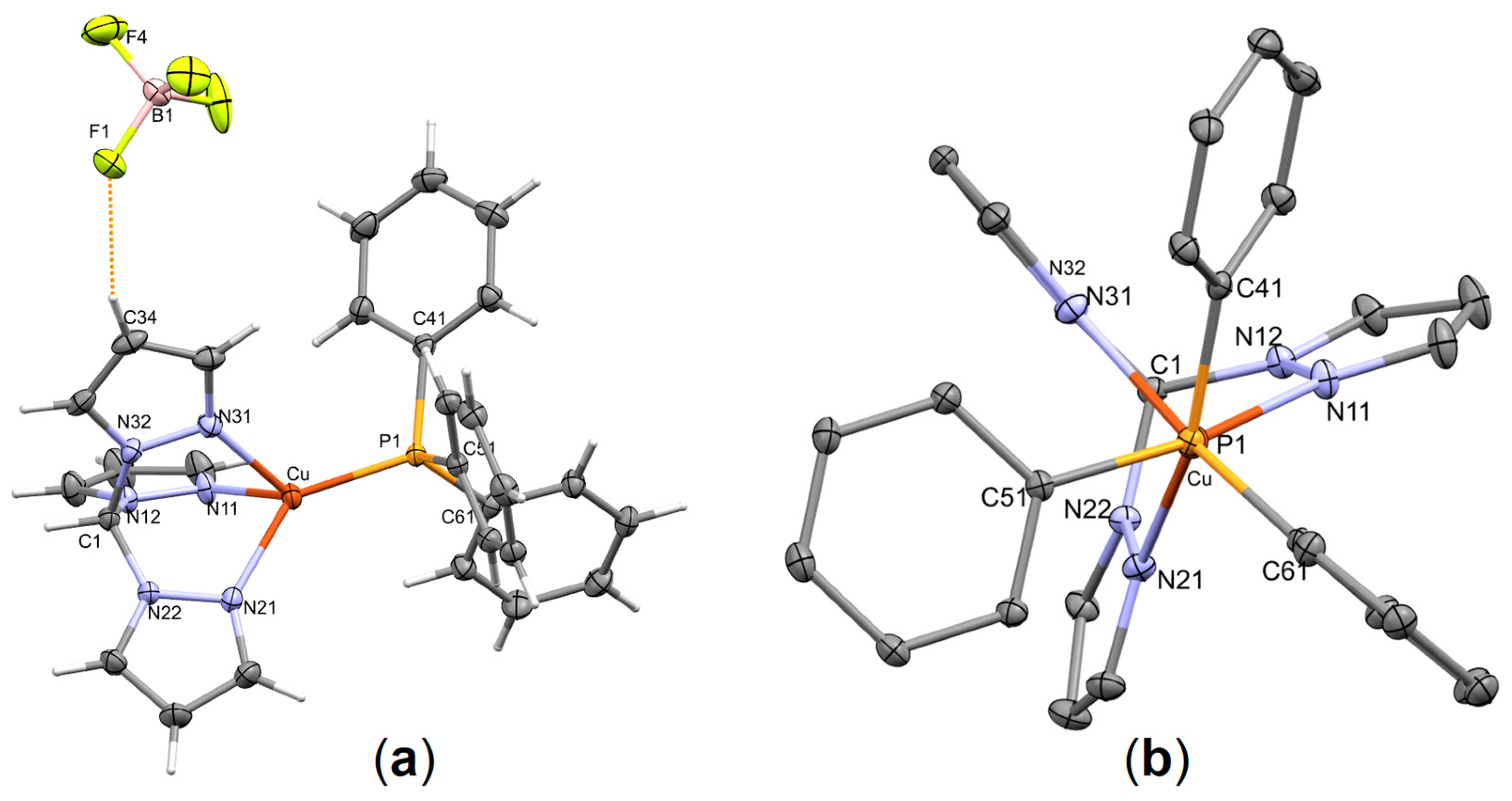
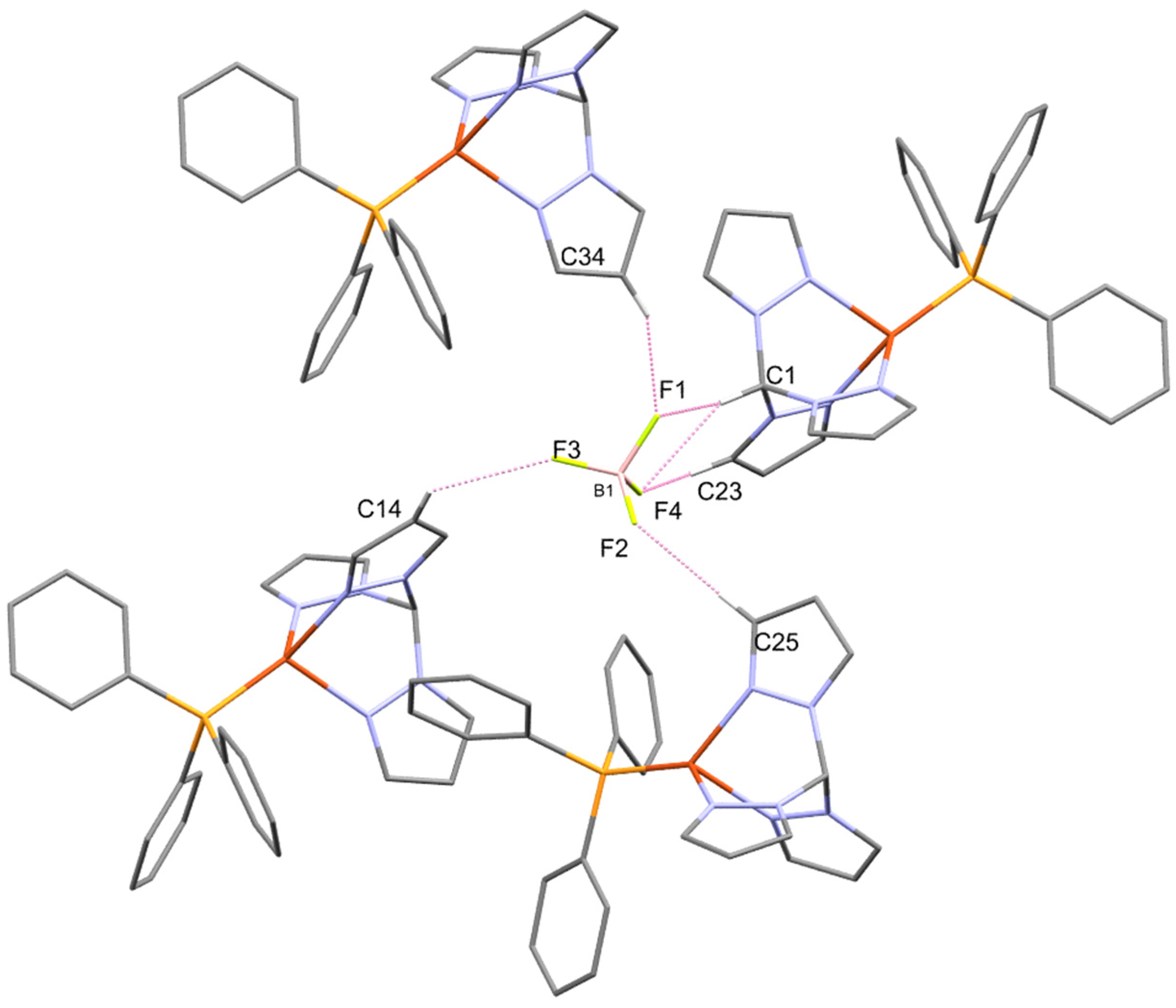
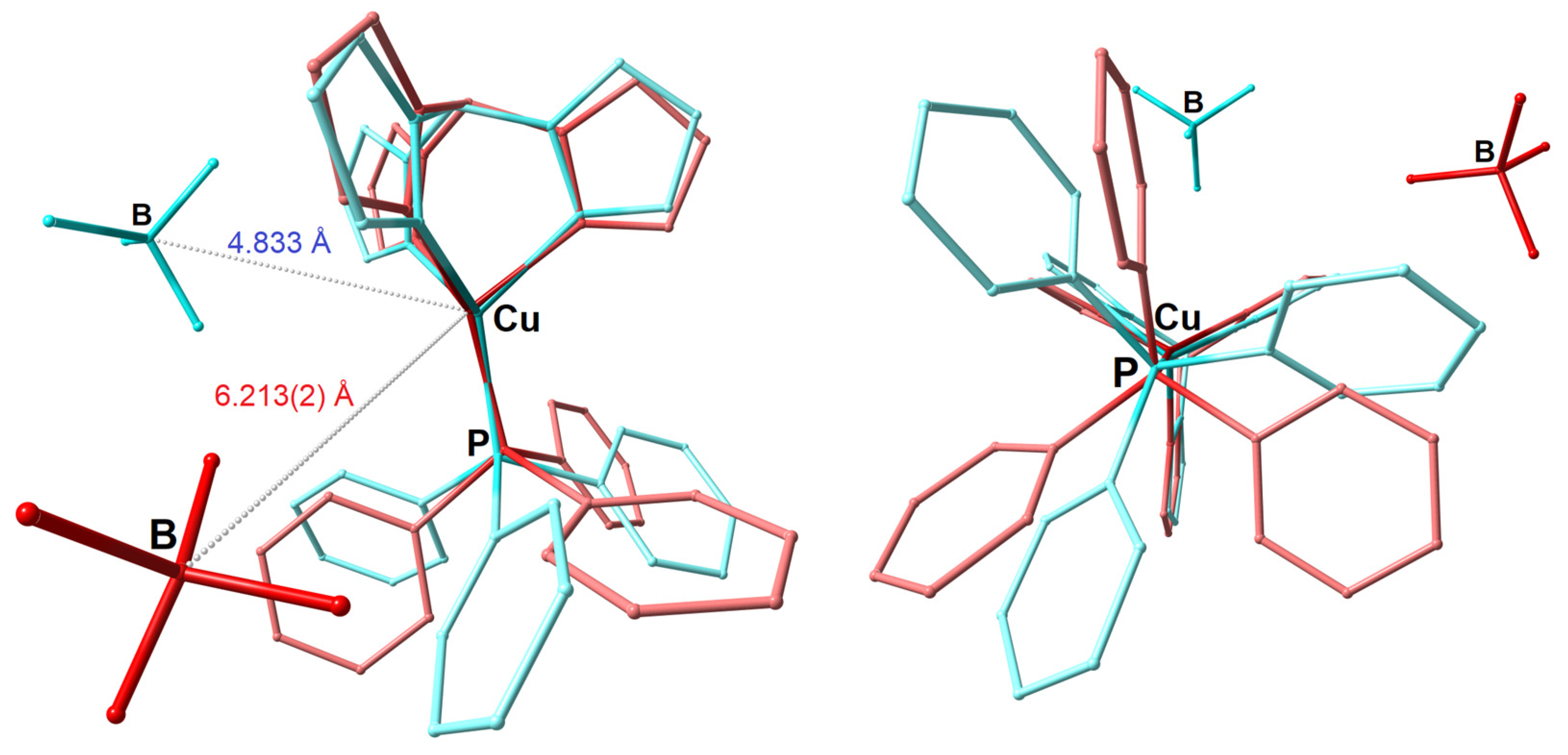
3.1. Synthesis and Single-Crystal X-ray Structure Determination
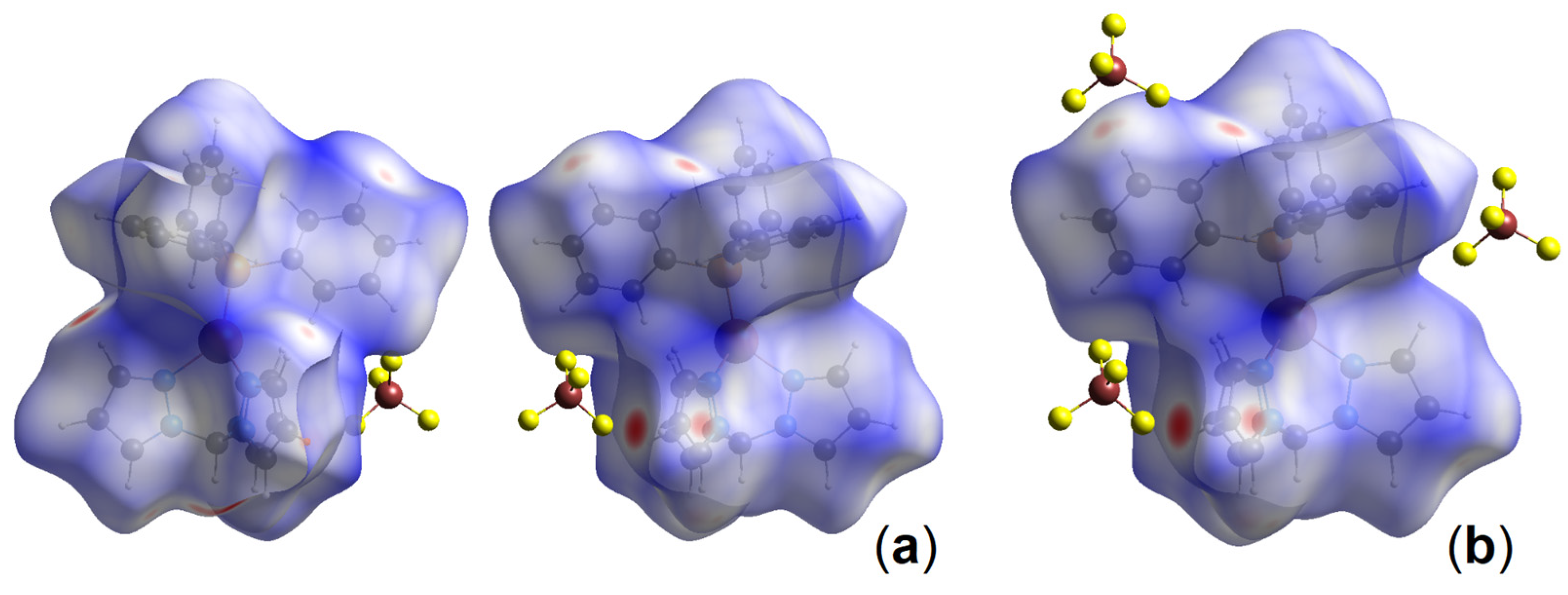
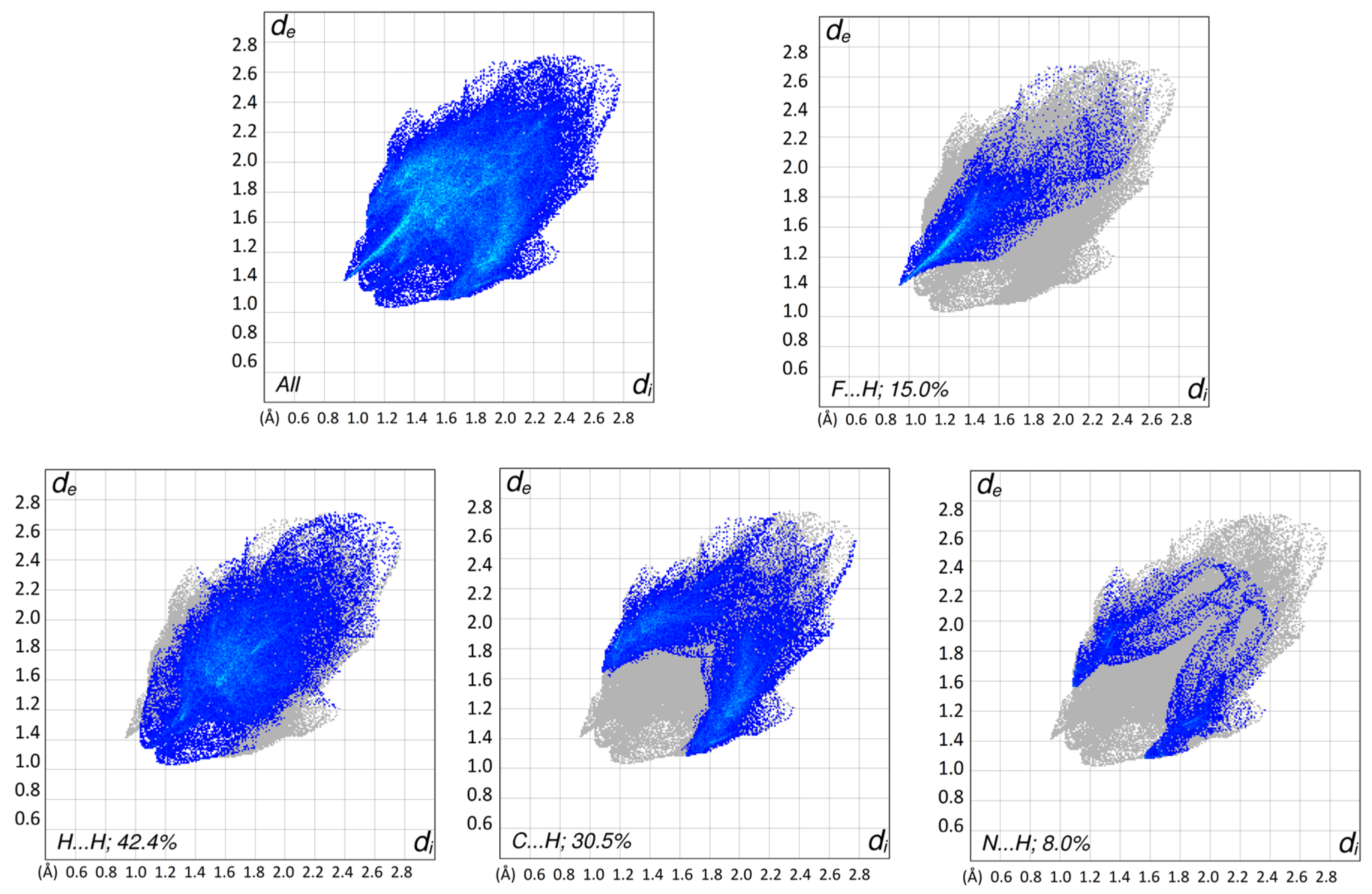
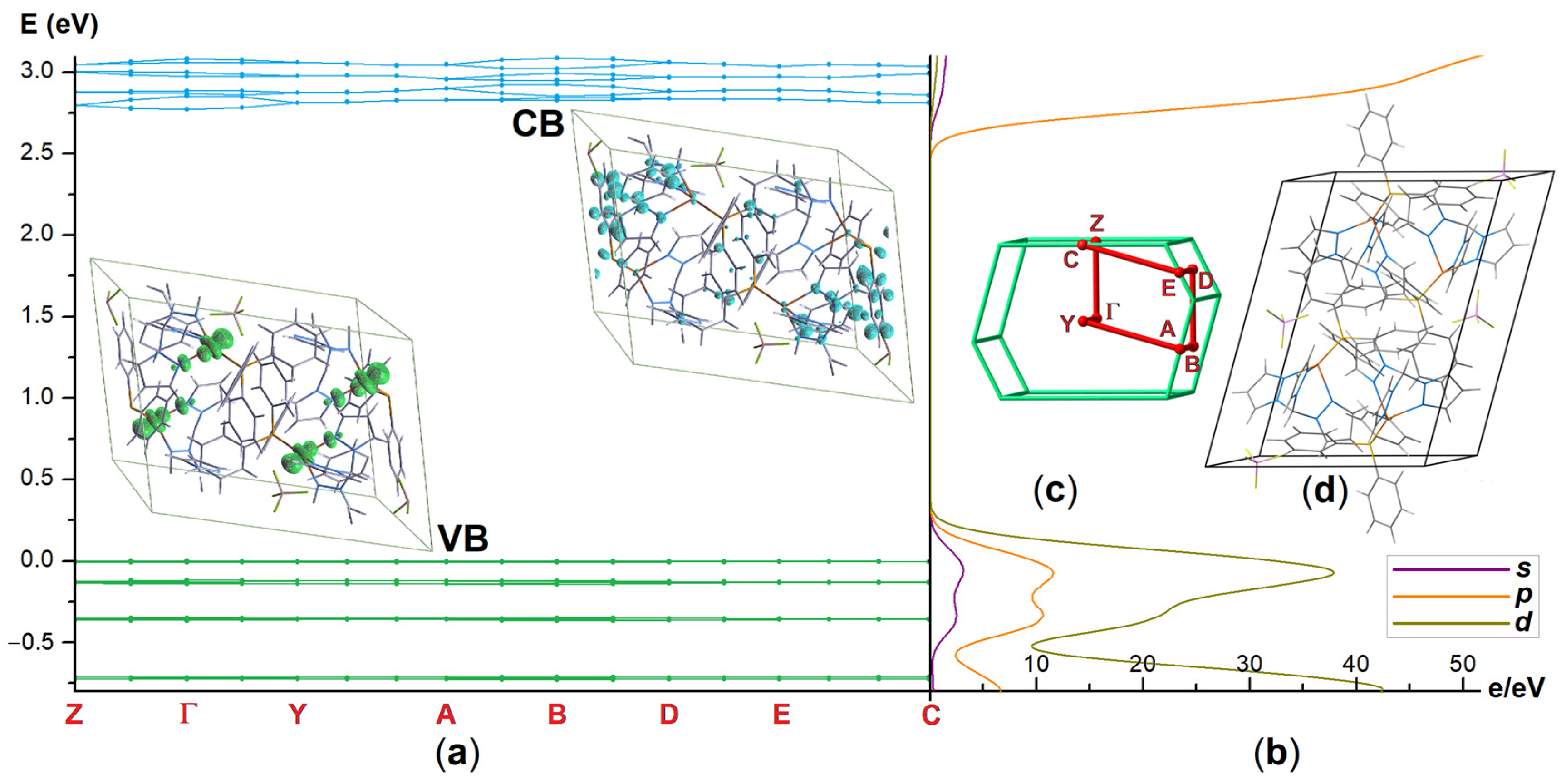
3.2. Hirshfeld Surface Analysis and Plane-Wave DFT Calculations
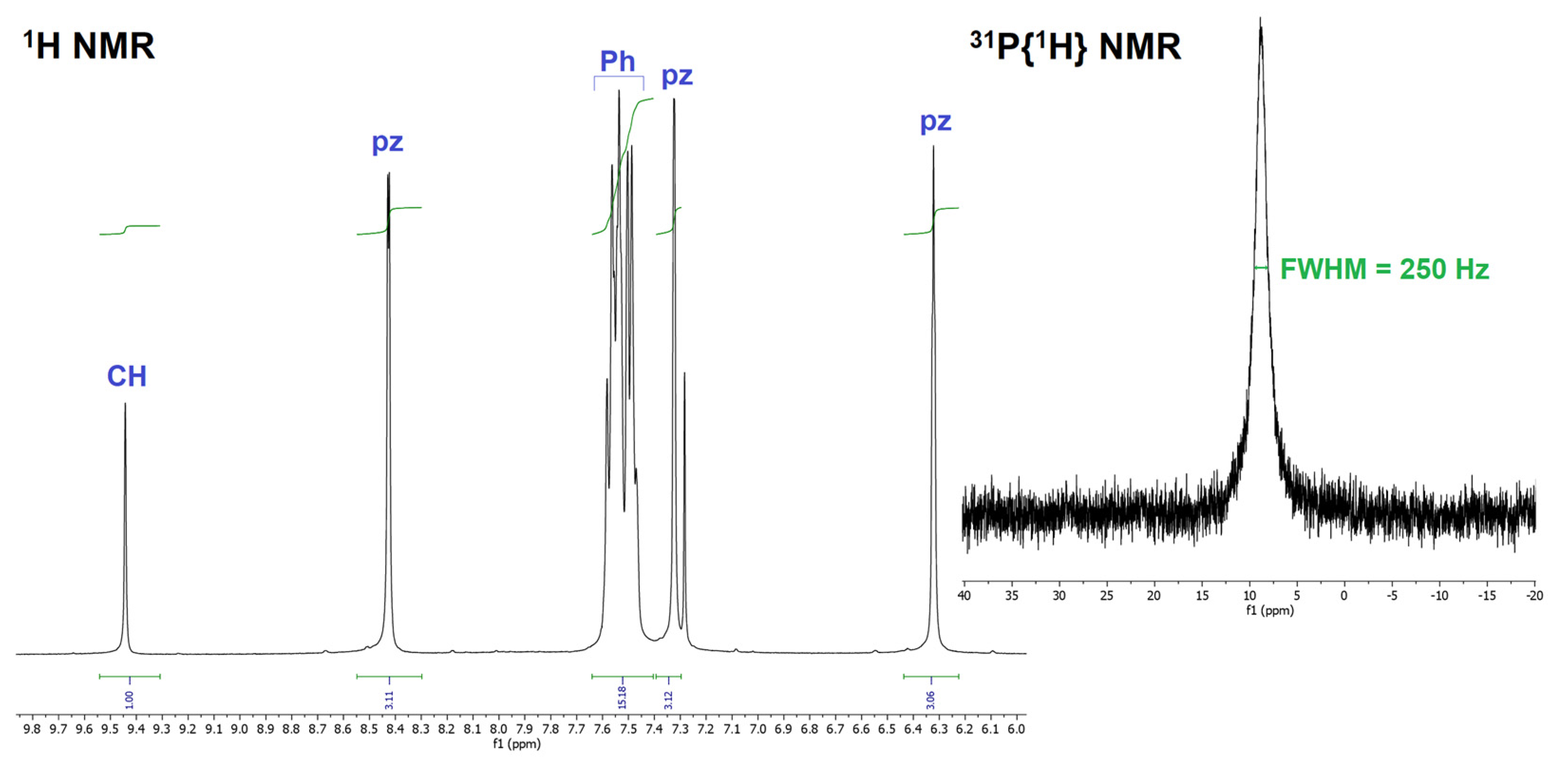
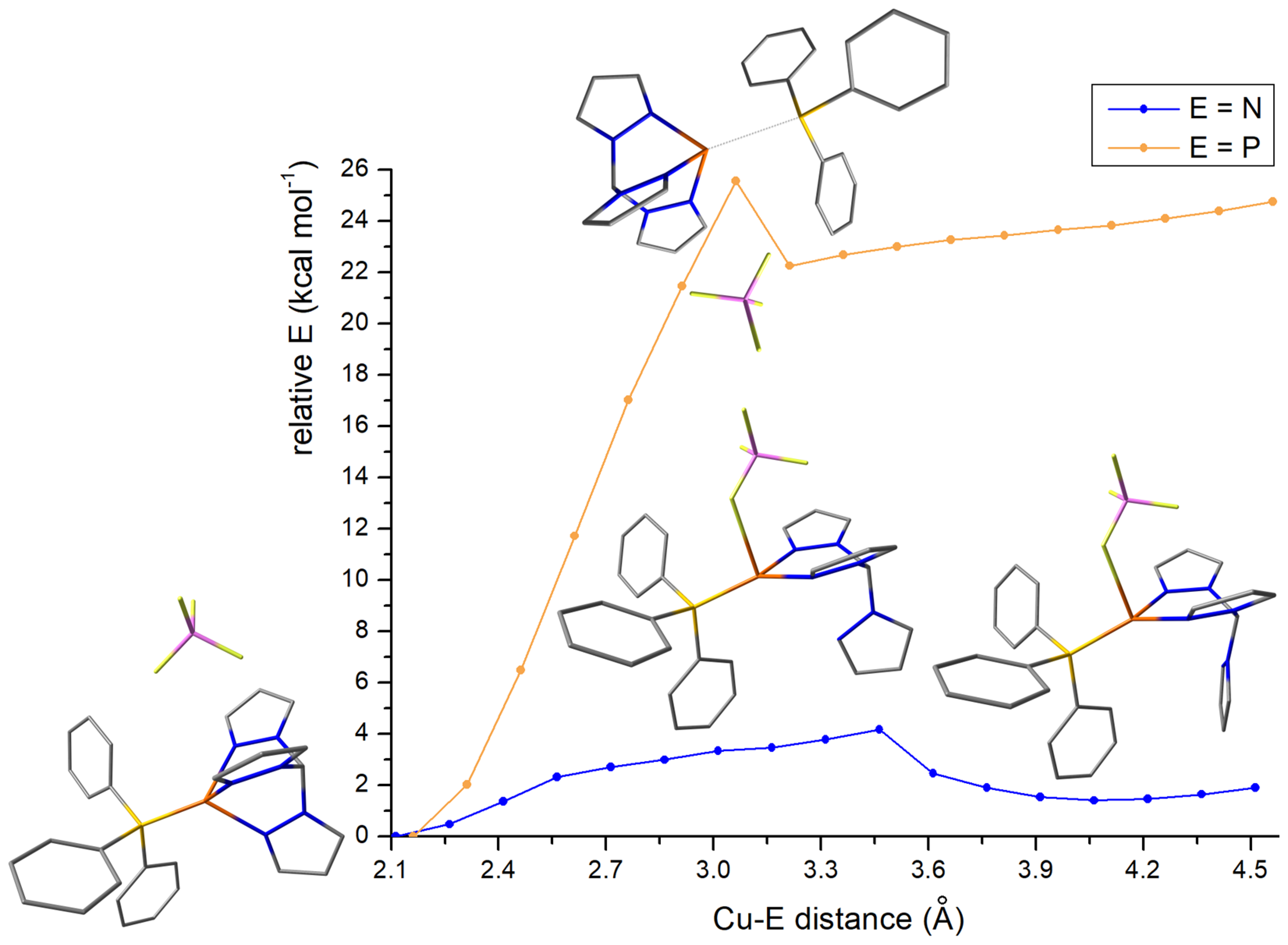
3.3. Fluxional Behaviour of the Complex in Solution
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reger, D.L. Tris(Pyrazolyl)Methane Ligands: The Neutral Analogs of Tris(Pyrazolyl)Borate Ligands. Comments Inorg. Chem. 1999, 21, 1–28. [Google Scholar] [CrossRef]
- Pettinari, C.; Pettinari, R. Metal derivatives of poly(pyrazolyl)alkanes I. Tris(pyrazolyl)alkanes and related systems. Coord. Chem. Rev. 2005, 249, 525–549. [Google Scholar] [CrossRef]
- Bigmore, H.R.; Lawrence, S.C.; Mountford, P.; Tredget, C.S. Coordination, organometallic and related chemistry of tris(pyrazolyl)methane ligands. Dalton Trans. 2005, 635–651. [Google Scholar] [CrossRef] [PubMed]
- Semeniuc, R.F.; Reger, D.L. Metal Complexes of Multitopic, Third Generation Poly(pyrazolyl)-methane Ligands: Multiple Coordination Arrangements. Eur. J. Inorg. Chem. 2016, 2016, 2253–2271. [Google Scholar] [CrossRef]
- Shakirova, O.G.; Lavrenova, L.G. Spin Crossover in New Iron(II) Coordination Compounds with Tris(pyrazol-1-yl)Methane. Crystals 2020, 10, 843. [Google Scholar] [CrossRef]
- Martins, L.M.D.R.S.; Pombeiro, A.J.L. Water-Soluble C-Scorpionate Complexes—Catalytic and Biological Applications. Eur. J. Inorg. Chem. 2016, 2016, 2236–2252. [Google Scholar] [CrossRef]
- McKeown, B.A.; Lee, J.P.; Mei, J.; Cundari, T.R.; Gunnoe, T.B. Transition Metal Mediated C–H Activation and Functionalization: The Role of Poly(pyrazolyl)borate and Poly(pyrazolyl)alkane Ligands. Eur. J. Inorg. Chem. 2016, 2016, 2296–2311. [Google Scholar] [CrossRef]
- Martins, L.M.D.R.S. C-Homoscorpionate Oxydation Catalysts–Electrochemical and Catalytic Activity. Catalysts 2017, 7, 12. [Google Scholar] [CrossRef]
- Mahmoud, A.G.; Martins, L.M.D.R.S.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Hydrosoluble Complexes Bearing Tris(pyrazolyl)methane Sulfonate Ligands: Synthesis, Characterization and Catalytic Activity for Henry Reaction. Catalysts 2019, 9, 611. [Google Scholar] [CrossRef]
- Cervinka, J.; Gobbo, A.; Biancalana, L.; Markova, L.; Novohradsky, V.; Guelfi, M.; Zacchini, S.; Kasparkova, J.; Brabec, V.; Marchetti, F. Ruthenium(II)–Tris-pyrazolylmethane Complexes Inhibit Cancer Cell Growth by Disrupting Mitochondrial Calcium Homeostasis. J. Med. Chem. 2022, 65, 10567–10587. [Google Scholar] [CrossRef]
- Gobbo, A.; Pereira, S.A.P.; Biancalana, L.; Zacchini, S.; Saraiva, M.L.M.F.S.; Dyson, P.J.; Marchetti, F. Anticancer ruthenium(II) tris(pyrazolyl)methane complexes with bioactive co-ligands. Dalton Trans. 2022, 51, 17050–17063. [Google Scholar] [CrossRef]
- Mani, F. Four-, five-, and six-co-ordinated iron(II) complexes with poly(1-pyrazolyl)methanes. Inorg. Nucl. Chem. Lett. 1979, 15, 297–302. [Google Scholar] [CrossRef]
- Muñoz-Molina, J.M.; Belderrain, T.R.; Pérez, P.J. Group 11 tris(pyrazolyl)methane complexes: Structural features and catalytic applications. Dalton Trans. 2019, 48, 10772–10781. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, E.R.; Mann, K.L.V.; Reeves, Z.R.; Behrendt, A.; Jeffery, J.C.; Maher, J.P.; McCleverty, J.A.; Ward, M.D. Copper(II) complexes of new potentially hexadentate N3S3- or N6-donor podand ligands based on the tris(pyrazolyl)borate or tris(pyrazolyl)methane core. New J. Chem. 1999, 23, 417–423. [Google Scholar] [CrossRef]
- Mesubi, M.A.; Anumba, F.O. Coordination Chemistry of Poly(1-pyrazolyl)alkanes, Part IV. Copper(II) Complexes of Bis- and Tris-(1-pyrazolyl)methane. Trans. Met. Chem. 1985, 10, 5–8. [Google Scholar] [CrossRef]
- Reger, D.L.; Collins, J.E.; Rheingold, A.L.; Liable-Sands, L.M. Synthesis and Characterization of Cationic [Tris(pyrazolyl)methane]copper(I) Carbonyl and Acetonitrile Complexes. Organometallics 1996, 15, 2029–2032. [Google Scholar] [CrossRef]
- Martini, D.; Pellei, M.; Pettinari, C.; Skelton, B.W.; White, A.H. Synthesis, spectroscopic and structural characterization of Cu(II) derivatives of tris(pyrazol-1-yl)methanes. Inorg. Chim. Acta 2002, 333, 72–82. [Google Scholar] [CrossRef]
- Melekhova, A.A.; Novikov, A.S.; Dubovtsev, A.Y.; Zolotarev, A.A.; Bokach, N.A. Tris(3,5-dimethylpyrazolyl)methane copper(I) complexes featuring one disubstituted cyanamide ligand. Inorg. Chim. Acta 2019, 484, 69–74. [Google Scholar] [CrossRef]
- Hsu, S.C.N.; Chen, H.H.Z.; Lin, I.-J.; Liu, J.-J.; Chen, P.-Y. Dinuclear copper(I) complexes of tris(3,5-dimethylpyrazol-1-yl)methane: Synthesis, structure, and reactivity. J. Organomet. Chem. 2007, 692, 3676–3684. [Google Scholar] [CrossRef]
- Fujisawa, K.; Tateda, A.; Miyashita, Y.; Okamoto, K.-i.; Paulat, F.; Praneeth, V.K.K.; Merkle, A.; Lehnert, N. Structural and Spectroscopic Characterization of Mononuclear Copper(I) Nitrosyl Complexes: End-on versus Side-on Coordination of NO to Copper(I). J. Am. Chem. Soc. 2008, 130, 1205–1218. [Google Scholar] [CrossRef]
- Kujime, M.; Izumi, C.; Tomura, M.; Hada, M.; Fujii, H. Effect of a Tridentate Ligand on the Structure, Electronic Structure, and Reactivity of the Copper(I) Nitrite Complex: Role of the Conserved Three-Histidine Ligand Environment of the Type-2 Copper Site in Copper-Containing Nitrite Reductases. J. Am. Chem. Soc. 2008, 130, 6088–6098. [Google Scholar] [CrossRef]
- Melekhova, A.A.; Novikov, A.S.; Bokach, N.A.; Avdonceva, M.S.; Kukushkin, V.Y. Characterization of Cu-ligand bonds in tris-pyrazolylmethane isocyanide copper(I) complexes based upon combined X-ray diffraction and theoretical study. Inorg. Chim. Acta 2016, 450, 140–145. [Google Scholar] [CrossRef]
- Porchia, M.; Dolmella, A.; Gandin, V.; Marzano, C.; Pellei, M.; Peruzzo, V.; Refosco, F.; Santini, C.; Tisato, F. Neutral and charged phosphine/scorpionate copper(I) complexes: Effects of ligand assembly on their antiproliferative activity. Eur. J. Med. Chem. 2013, 59, 218–226. [Google Scholar] [CrossRef]
- Fujisawa, K.; Noguchi, Y.; Miyashita, Y.; Okamoto, K.-i.; Lehnert, N. Mononuclear and Binuclear Copper(I) Complexes Ligated by Bis(3,5-diisopropyl-1-pyrazolyl)methane: Insight into the Fundamental Coordination Chemistry of Three-Coordinate Copper(I) Complexes with a Neutral Coligand. Inorg. Chem. 2007, 46, 10607–10623. [Google Scholar] [CrossRef]
- Pellei, M.; Lobbia, G.G.; Santini, C.; Spagna, R.; Camalli, M.; Fedeli, D.; Falcioni, G. Synthesis, characterization and antioxidant activity of new copper(I) complexes of scorpionate and water soluble phosphane ligands. Dalton Trans. 2004, 2822–2828. [Google Scholar] [CrossRef]
- Wanke, R.; Smolenski, P.; Guedes Da Silvan, M.F.C.; Martins, L.M.D.R.S.; Pombeiro, A.J. Cu(I) complexes bearing the new sterically demanding and coordination flexible tris(3-phenyl-1-pyrazolyl)methanesulfonate ligand and the water-soluble phosphine 1,3,5-triaza-7-phosphaadamantane or related ligands. Inorg. Chem. 2008, 47, 10158–10168. [Google Scholar] [CrossRef]
- Choi, I.Y.; Ahn, S.; Seo, J.; Park, K.-M. Synthesis and Structural Characterization of an Air-stable {[HC(pyrazolyl)3]Cu(PPh3)}PF6. Bull. Korean Chem. Soc. 2004, 25, 1065–1067. [Google Scholar] [CrossRef]
- Reger, D.L.; Semeniuc, R.F.; Smith, M.D. Supramolecular structures of tris(pyrazolyl)methane complexes of triphenylphosphine copper(I). Rev. Roum. Chim. 2002, 47, 1037–1046. [Google Scholar]
- Ferraro, V.; Castro, J.; Agostinis, L.; Bortoluzzi, M. Luminescent heteroleptic copper(I) complexes with polydentate benzotriazolyl-based ligands. Trans. Met. Chem. 2021, 46, 391–402. [Google Scholar] [CrossRef]
- Ferraro, V.; Bortoluzzi, M.; Castro, J. Synthesis of Bis(benzotriazol-1-yl)methane Derivatives by Cobalt-Catalyzed Formation of C-C Bonds. Chem. Proc. 2019, 41, 29. [Google Scholar] [CrossRef]
- Ferraro, V.; Bortoluzzi, M.; Castro, J.; Vomiero, A.; You, S. Luminescent Cu(I) complex with bis(indazol-1-yl)phenylmethane as chelating ligand. Inorg. Chem. Commun. 2020, 116, 107894. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C.L.L. Purification of Laboratory Chemicals, 5th ed.; Butterworth-Heinemann: London, UK, 2003. [Google Scholar]
- Kubas, G.J.; Monzyk, B.; Crumblis, A.L. Tetrakis(Acetonitrile)Copper(1+) hexafluorophosphate(1-). Inorg. Synth. 1990, 28, 68–70. [Google Scholar] [CrossRef]
- Reger, D.L.; Grattan, T.C.; Brown, K.J.; Little, C.A.; Lamba, J.J.S.; Rheingold, A.L.; Sommer, R.D. Syntheses of tris(pyrazolyl)methane ligands and {[tris(pyrazolyl)methane]Mn(CO)3}SO3CF3 complexes: Comparison of ligand donor properties. J. Organomet. Chem. 2000, 607, 120–128. [Google Scholar] [CrossRef]
- Bruker AXS Inc. APEX3, SMART, SAINT; Bruker AXS Inc.: Madison, WI, USA, 2015. [Google Scholar]
- McArdle, P. Oscail, a program package for small-molecule single-crystal crystallography with crystal morphology prediction and molecular modelling. J. Appl. Crystallogr. 2017, 50, 320–326. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Spek, A.L. checkCIF validation ALERTS: What they mean and how to respond. Acta Crystallogr. 2020, E76, 1–11. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Cryst. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]
- Wolff, S.K.; Grimwood, D.J.; McKinnon, J.J.; Turner, M.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer, Version 3.1. University of Western Australia: Perth, Australia. Available online: www.crystalexplorer.net (accessed on 16 January 2024).
- Spackman, M.A.; MacKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef]
- Lin, J.S.; Qteish, A.; Payne, M.C.; Heine, V. Optimized and transferable nonlocal separable ab initio pseudopotentials. Phys. Rev. B 1993, 47, 4174–4180. [Google Scholar] [CrossRef]
- Tkatchenko, A.; Scheffler, M. Accurate Molecular Van Der Waals Interactions from Ground-State Electron Density and Free-Atom Reference Data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef]
- Koelling, D.D.; Harmon, B.N. A technique for relativistic spin-polarised calculations. J. Phys. C Solid State Phys. 1977, 10, 3107–3114. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Kristallogr. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Rutter, M.J. C2x: A tool for visualisation and input preparation for Castep and other electronic structure codes. Comput. Phys. Commun. 2018, 225, 174–179. [Google Scholar] [CrossRef]
- Hinuma, Y.; Pizzi, G.; Kumagai, Y.; Oba, F.; Tanaka, I. Band structure diagram paths based on crystallography. Comput. Mater. Sci. 2017, 128, 140–184. [Google Scholar] [CrossRef]
- Kokalj, A. Computer graphics and graphical user interfaces as tools in simulations of matter at the atomic scale. Comp. Mater. Sci. 2003, 28, 155–168. [Google Scholar] [CrossRef]
- Kokalj, A. XCrySDen—A new program for displaying crystalline structures and electron densities. J. Mol. Graph. Model. 1999, 17, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Hansen, A.; Ehlert, S.; Mewes, J.-M. r2SCAN-3c: A “Swiss army knife” composite electronic-structure method. J. Chem. Phys. 2021, 154, 064103. [Google Scholar] [CrossRef] [PubMed]
- Furness, J.W.; Kaplan, A.D.; Ning, J.; Perdew, J.P.; Sun, J. Accurate and Numerically Efficient r2SCAN Meta-Generalized Gradient Approximation. J. Phys. Chem. Lett. 2020, 11, 8208–8215. [Google Scholar] [CrossRef] [PubMed]
- Kruse, H.; Grimme, S. A geometrical correction for the inter- and intra-molecular basis set superposition error in Hartree-Fock and density functional theory calculations for large systems. J. Chem. Phys. 2012, 136, 154101. [Google Scholar] [CrossRef] [PubMed]
- Caldeweyher, E.; Bannwarth, C.; Grimme, S. Extension of the D3 dispersion coefficient model. J. Chem. Phys. 2017, 147, 034112. [Google Scholar] [CrossRef] [PubMed]
- Caldeweyher, E.; Ehlert, S.; Hansen, A.; Neugebauer, H.; Spicher, S.; Bannwarth, C.; Grimme, S. A generally applicable atomic-charge dependent London dispersion correction. J. Chem. Phys. 2019, 150, 154122. [Google Scholar] [CrossRef] [PubMed]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system-Version 5.0. WIREs Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Geary, W.J. The use of conductivity measurements in organic solvents for the characterisation of coordination compounds. Coord. Chem. Rev. 1971, 7, 81–122. [Google Scholar] [CrossRef]
- Cirera, J.; Alemany, P.; Alvarez, S. Mapping the Stereochemistry and Symmetry of Tetracoordinate Transition-Metal Complexes. Chem. Eur. J. 2004, 10, 190–207. [Google Scholar] [CrossRef]
- Alvarez, S.; Alemany, P.; Casanova, D.; Cirera, J.; Llunell, M.; Avnir, D. Shape maps and polyhedral interconversion paths in transition metal chemistry. Coord. Chem. Rev. 2005, 249, 1693–1708. [Google Scholar] [CrossRef]
- Yang, L.; Powell, D.R.; Houser, R.P. Structural variation in copper(I) complexes with pyridylmethylamide ligands: Structural analysis with a new four-coordinate geometry index, τ4. Dalton Trans. 2007, 955–964. [Google Scholar] [CrossRef]
- Okuniewski, A.; Rosiak, D.; Chojnacki, J.; Becker, B. Coordination polymers and molecular structures among complexes of mercury(II) halides with selected 1-benzoylthioureas. Polyhedron 2015, 90, 47–57. [Google Scholar] [CrossRef]
- MacKinnon, J.J.; Spackman, M.A.; Mitchell, A.S. Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2004, 60, 627–668. [Google Scholar] [CrossRef] [PubMed]
- Legon, A.C.; Walker, N.R. What’s in a name? ‘Coinage-metal’ non-covalent bonds and their definition. Chem. Phys. Phys. Chem. 2018, 20, 19332–19338. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Zhang, Y.; Tian, J.; Liu, Z. Aquabis-(triphenyl-phosphine-kP)copper(I) tetrafluoridoborate. Acta Crystallogr. 2009, E65, m1001. [Google Scholar] [CrossRef]
- Titov, A.A.; Filippov, O.A.; Smol’yakov, A.F.; Averin, A.A.; Shubina, E.S. Copper(I) complex with BINAP and 3,5-dimethylpyrazole: Synthesis and photoluminescent properties. Mendeleev Commun. 2019, 29, 570–572. [Google Scholar] [CrossRef]
- Ferraro, V.; Girotto, M.; Castro, J.; Bortoluzzi, M. Intense millisecond-long red luminescence from heteroleptic Cu(I) 2,1,3-benzothiadiazole complexes. Dye. Pigment. 2023, 217, 111388. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CCDC code | 2323499 |
| Empirical formula | C28H25BCuF4N6P |
| Moiety formula | C28H25CuN6P, BF4 |
| Formula weight | 626.86 |
| Temperature | 100(2) K |
| Wavelength | 0.71073 Å |
| Crystal system | Monoclinic |
| Space group | P21/c |
| Unit cell dimensions | a = 11.9442(9) Å |
| b = 15.5313(9) Å | |
| c = 15.9059(12) Å | |
| β = 105.945(3)° | |
| Volume | 2837.2(3) Å3 |
| Z | 4 |
| Density (calculated) | 1.468 Mg/m3 |
| Absorption coefficient | 0.882 mm−1 |
| F(000) | 1280 |
| Crystal size | 0.273 × 0.251 × 0.219 mm |
| Theta range for data collection | 2.623 to 28.282° |
| Index ranges | −15 ≤ h ≤ 13 |
| −20 ≤ k ≤ 20 | |
| −21 ≤ l ≤ 21 | |
| Reflections collected | 80174 |
| Independent reflections | 7023 [Rint = 0.0322] |
| Reflections observed (>2σ) | 6568 |
| Data Completeness | 0.999 |
| Absorption correction | Semi-empirical from equivalents |
| Max. and min. transmission | 0.7457 and 0.6079 |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 7023/0/370 |
| Goodness-of-fit on F2 | 1.062 |
| Final R indices [I > 2σ(I)] | R1 = 0.0272 |
| wR2 = 0.0667 | |
| R indices (all data) | R1 = 0.0297 |
| wR2 = 0.0680 | |
| Largest diff. peak and hole | 0.761 and −0.485 e.Å−3 |
| Anion | SP-4 1 | T-4 2 | SS-4 3 | vTBPY-4 4 | τ4 |
|---|---|---|---|---|---|
| [BF4]− | 32.393 | 4.329 | 7.120 | 3.887 | 0.67 |
| [PF6]− | 31.757 | 4.288 | 6.741 | 3.937 | 0.67 |
| [NO3]− | 32.431 | 4.173 | 6.885 | 4.702 | 0.72 |
| D-H…A | d(D-H) | d(H…A) | d(D…A) | <(DHA) |
|---|---|---|---|---|
| C(34)-H(34)…F(1) | 0.95 | 2.27 | 3.1367(18) | 151.6 |
| C(1)-H(1)…F(1 i) | 1.00 | 2.26 | 3.1381(16) | 146.5 |
| C(13)-H(13)…F(1 i) | 0.95 | 2.42 | 3.1223(19) | 130.7 |
| C(25)-H(25)…F(2 ii) | 0.95 | 2.36 | 3.3073(17) | 174.7 |
| C(14)-H(14)…F(3 iii) | 0.95 | 2.49 | 3.342(2) | 149.0 |
| C(23)-H(23)…F(4 i) | 0.95 | 2.53 | 3.2634(19) | 133.9 |
| C(1)-H(1)…F(4 i) | 1.00 | 2.45 | 3.3596(18) | 150.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castro, J.; Ferraro, V.; Bortoluzzi, M. Single-Crystal X-ray Structure Determination of Tris(pyrazol-1-yl)methane Triphenylphosphine Copper(I) Tetrafluoroborate, Hirshfeld Surface Analysis and DFT Calculations. Crystals 2024, 14, 162. https://doi.org/10.3390/cryst14020162
Castro J, Ferraro V, Bortoluzzi M. Single-Crystal X-ray Structure Determination of Tris(pyrazol-1-yl)methane Triphenylphosphine Copper(I) Tetrafluoroborate, Hirshfeld Surface Analysis and DFT Calculations. Crystals. 2024; 14(2):162. https://doi.org/10.3390/cryst14020162
Chicago/Turabian StyleCastro, Jesús, Valentina Ferraro, and Marco Bortoluzzi. 2024. "Single-Crystal X-ray Structure Determination of Tris(pyrazol-1-yl)methane Triphenylphosphine Copper(I) Tetrafluoroborate, Hirshfeld Surface Analysis and DFT Calculations" Crystals 14, no. 2: 162. https://doi.org/10.3390/cryst14020162