
Polymer-Mediated Delivery of siRNAs to Hepatocellular Carcinoma: Variables Affecting Specificity and Effectiveness


 , ,
, ,  , , , , ,
, , , , , 
Abstract
:1. Introduction
1.1. HCC
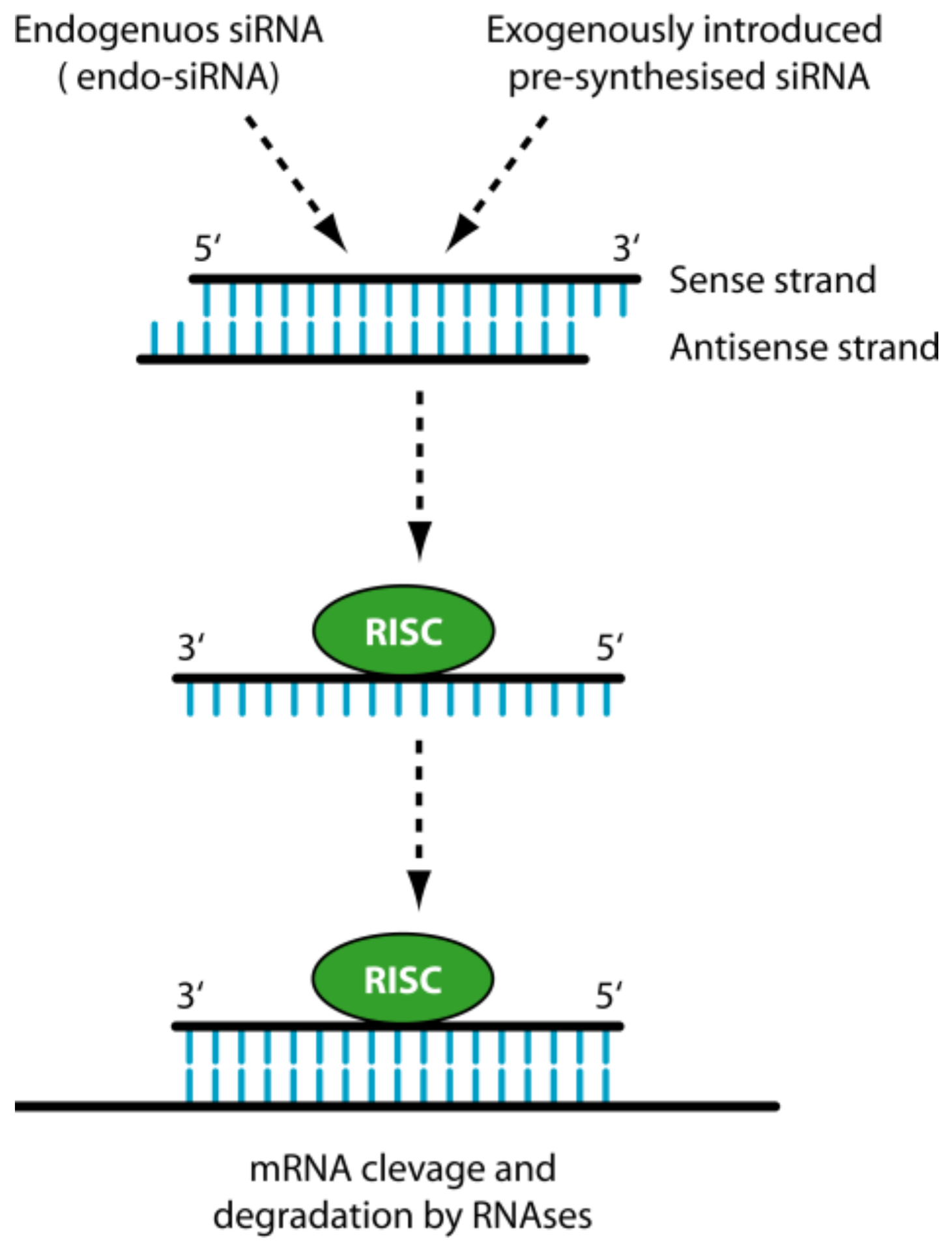
1.2. siRNA
2. The Delivery Problems of siRNAs
3. General Strategies to Optimize siRNA Delivery
3.1. Polymers for siRNA Delivery
4. Specific Strategies to Optimize siRNA Delivery to HCC
4.1. Vascular Aspects
4.2. Phagocytosis
4.3. Physical Aspects
4.4. Molecular Aspects: Targeting Surface Antigens of HCC Cells
4.5. Molecular Aspects: Targeting HCC Specific Oncogenes
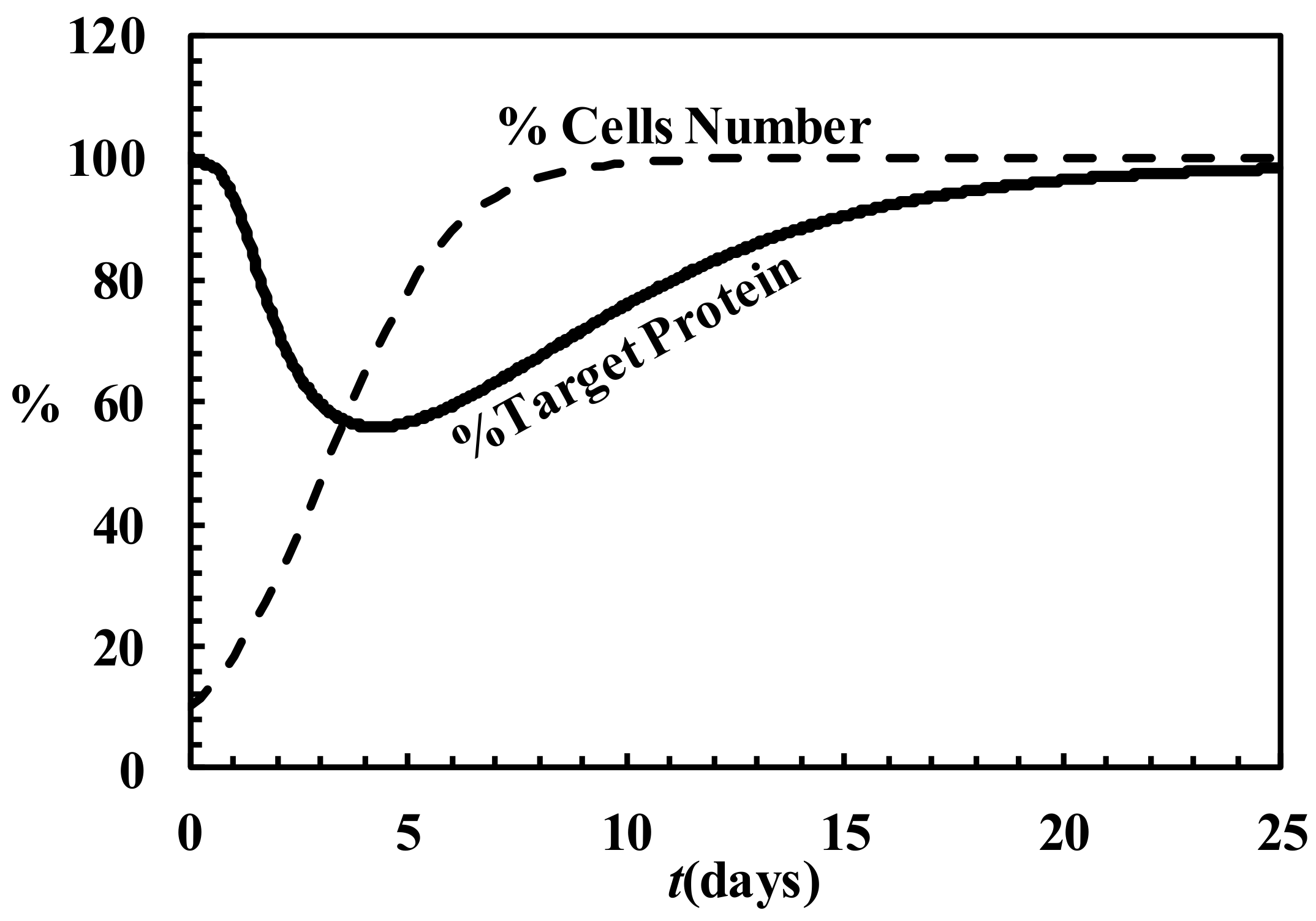
4.6. Description of siRNA Activity by Mathematical Modeling

5. Strategies Utilized to Deliver siRNA to HCC
5.1. In Vitro Models of HCC
5.2. In Vivo Models of HCC
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Grassi, G.; Marini, J.C. Ribozymes: Structure, function, and potential therapy for dominant genetic disorders. Ann. Med. 1996, 28, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Grassi, G.; Dawson, P.; Guarnieri, G.; Kandolf, R.; Grassi, M. Therapeutic potential of hammerhead ribozymes in the treatment of hyper-proliferative diseases. Curr. Pharm. Biotechnol. 2004, 5, 369–386. [Google Scholar] [CrossRef] [PubMed]
- Agostini, F.; Dapas, B.; Farra, R.; Grassi, M.; Racchi, G.; Klingel, K.; Kandolf, R.; Heidenreich, O.; Mercatahnti, A.; Rainaldi, G.; et al. Potential applications of small interfering RNAs in the cardiovascular field. Drug Future 2006, 31, 513–525. [Google Scholar] [CrossRef]
- Grassi, M.; Cavallaro, G.; Scirè, S.; Scaggiante, B.; Daps, B.; Farra, R.; Baiz, D.; Giansante, C.; Guarnieri, G.; Perin, D.; et al. Current Strategies to Improve the Efficacy and the Delivery of Nucleic Acid Based Drugs. Curr. Signal Transduct. Ther. 2010, 5, 92–120. [Google Scholar] [CrossRef]
- Grassi, G.; Schneider, A.; Engel, S.; Racchi, G.; Kandolf, R.; Kuhn, A. Hammerhead ribozymes targeted against cyclin E and E2F1 cooperate to down-regulate coronary smooth muscle cell proliferation. J. Gene Med. 2005, 7, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Palazzolo, S.; Bayda, S.; Corona, G.; Toffoli, G.; Rizzolio, F. DNA Nanotechnology for Cancer Therapy. Theranostics 2016, 6, 710–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, Z.D. Neoplasms of the liver. Mod. Pathol. 2007, 20 (Suppl. 1), S49–S60. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Knox, J.J.; Cleary, S.P.; Dawson, L.A. Localized and systemic approaches to treating hepatocellular carcinoma. J. Clin. Oncol. 2015, 33, 1835–1844. [Google Scholar] [CrossRef] [PubMed]
- Venook, A.P.; Papandreou, C.; Furuse, J.; de Guevara, L.L. The incidence and epidemiology of hepatocellular carcinoma: A global and regional perspective. Oncologist 2010, 15 (Suppl. 4), 5–13. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Millonig, G.; Seitz, H.K. Alcoholic liver disease and hepatitis C: A frequently underestimated combination. World J. Gastroenterol. 2009, 15, 3462–3471. [Google Scholar] [PubMed]
- Llovet, J.M.; Burroughs, A.; Bruix, J. Hepatocellular carcinoma. Lancet 2003, 362, 1907–1917. [Google Scholar] [CrossRef]
- Dhanasekaran, R.; Limaye, A.; Cabrera, R. Hepatocellular carcinoma: Current trends in worldwide epidemiology, risk factors, diagnosis, and therapeutics. Hepat. Med. 2012, 4, 19–37. [Google Scholar] [PubMed]
- El-Serag, H.B. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012, 142, 1264–1273. [Google Scholar] [CrossRef] [PubMed]
- Schlachterman, A.; Craft, W.W., Jr.; Hilgenfeldt, E.; Mitra, A.; Cabrera, R. Current and future treatments for hepatocellular carcinoma. World J. Gastroenterol. 2015, 21, 8478–8491. [Google Scholar] [CrossRef] [PubMed]
- Lencioni, R.; Marrero, J.; Venook, A.; Ye, S.L.; Kudo, M. Design and rationale for the non-interventional Global Investigation of Therapeutic DEcisions in Hepatocellular Carcinoma and Of its Treatment with Sorafenib (GIDEON) study. Int. J. Clin. Pract. 2010, 64, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Gabrielson, A.; Tesfaye, A.A.; Marshall, J.L.; Pishvaian, M.J.; Smaglo, B.; Jha, R.; Dorsch-Vogel, K.; Wang, H.; He, A.R. Phase II study of temozolomide and veliparib combination therapy for sorafenib- refractory advanced hepatocellular carcinoma. Cancer Chemother. Pharmacol. 2015, 76, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Sherman, M. Management of hepatocellular carcinoma: An update. Hepatology 2011, 53, 1020–1022. [Google Scholar] [CrossRef] [PubMed]
- Ryder, S.D. Guidelines for the diagnosis and treatment of hepatocellular carcinoma (HCC) in adults. Gut 2003, 52 (Suppl. 3), iii1–iii8. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Bruix, J. Systematic review of randomized trials for unresectable hepatocellular carcinoma: Chemoembolization improves survival. Hepatology 2003, 37, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef]
- Scaggiante, B.; Dapas, B.; Farra, R.; Grassi, M.; Pozzato, G.; Giansante, C.; Fiotti, N.; Grassi, G. Improving siRNA bio-distribution and minimizing side effects. Curr. Drug Metab. 2011, 12, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Werth, D.; Grassi, G.; Konjer, N.; Dapas, B.; Farra, R.; Giansante, C.; Kandolf, R.; Guarnieri, G.; Nordheim, A.; Heidenreich, O. Proliferation of human primary vascular smooth muscle cells depends on serum response factor. Eur. J. Cell Biol. 2010, 89, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Dapas, B.; Farra, R.; Grassi, M.; Giansante, C.; Fiotti, N.; Uxa, L.; Rainaldi, G.; Mercatanti, A.; Colombatti, A.; Spessotto, P.; et al. Role of E2F1-cyclin E1-cyclin E2 circuit in human coronary smooth muscle cell proliferation and therapeutic potential of its downregulation by siRNAs. Mol. Med. 2009, 15, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Farra, R.; Grassi, M.; Grassi, G.; Dapas, B. Therapeutic potential of small interfering RNAs/micro interfering RNA in hepatocellular carcinoma. World J. Gastroenterol. 2015, 21, 8994–9001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farra, R.; Dapas, B.; Baiz, D.; Tonon, F.; Chiaretti, S.; Del, S.G.; Rustighi, A.; Elvassore, N.; Pozzato, G.; Grassi, M.; et al. Impairment of the Pin1/E2F1 axis in the anti-proliferative effect of bortezomib in hepatocellular carcinoma cells. Biochimie 2015, 112, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Farra, R.; Dapas, B.; Pozzato, G.; Scaggiante, B.; Agostini, F.; Zennaro, C.; Grassi, M.; Rosso, N.; Giansante, C.; Fiotti, N.; et al. Effects of E2F1-cyclin E1-E2 circuit down regulation in hepatocellular carcinoma cells. Dig. Liver Dis. 2011, 43, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Farra, R.; Dapas, B.; Pozzato, G.; Giansante, C.; Heidenreich, O.; Uxa, L.; Zennaro, C.; Guarnieri, G.; Grassi, G. Serum response factor depletion affects the proliferation of the hepatocellular carcinoma cells HepG2 and JHH6. Biochimie 2010, 92, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Hong, J.; Zheng, S.; Ding, Y.; Guo, S.; Zhang, H.; Zhang, X.; Du, Q.; Liang, Z. Elimination pathways of systemically delivered siRNA. Mol. Ther. 2011, 19, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.L.; Linsley, P.S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010, 9, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, Y. Gene therapy: A battle against biological barriers. Curr. Mol. Med. 2001, 1, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C.; Rizzolio, F.; Bordoni, V.; Caligiuri, I.; Manera, C.; Macchia, M.; Minutolo, F.; Martinelli, A.; Giordano, A.; Tuccinardi, T. 4-Aryliden-2-methyloxazol-5(4H)-one as a new scaffold for selective reversible MAGL inhibitors. J. Enzyme Inhib. Med. Chem. 2016, 31, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Tuccinardi, T.; Granchi, C.; Rizzolio, F.; Caligiuri, I.; Battistello, V.; Toffoli, G.; Minutolo, F.; Macchia, M.; Martinelli, A. Identification and characterization of a new reversible MAGL inhibitor. Bioorg. Med. Chem. 2014, 22, 3285–3291. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Tuccinardi, T.; Rizzolio, F.; Caligiuri, I.; Botta, L.; Granchi, C.; Ortore, G.; Minutolo, F.; Schenone, S.; Martinelli, A. Identification of new Fyn kinase inhibitors using a FLAP-based approach. J. Chem. Inf. Model. 2013, 53, 2538–2547. [Google Scholar] [CrossRef] [PubMed]
- Manera, C.; Saccomanni, G.; Malfitano, A.M.; Bertini, S.; Castelli, F.; Laezza, C.; Ligresti, A.; Lucchesi, V.; Tuccinardi, T.; Rizzolio, F.; et al. Rational design, synthesis and anti-proliferative properties of new CB2 selective cannabinoid receptor ligands: An investigation of the 1,8-naphthyridin-2(1H)-one scaffold. Eur. J. Med. Chem. 2012, 52, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Bayda, S.; Hadla, M.; Caligiuri, I.; Russo, S.C.; Palazzolo, S.; Kempter, S.; Corona, G.; Toffoli, G.; Rizzolio, F. Enhanced Chemotherapeutic Behavior of Open-Caged DNA@Doxorubicin Nanostructures for Cancer Cells. J. Cell. Physiol. 2016, 231, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Toffoli, G.; Hadla, M.; Corona, G.; Caligiuri, I.; Palazzolo, S.; Semeraro, S.; Gamini, A.; Canzonieri, V.; Rizzolio, F. Exosomal doxorubicin reduces the cardiac toxicity of doxorubicin. Nanomedicine (Lond.) 2015, 10, 2963–2971. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Toffoli, G.; Rizzolio, F. Fluorescent carbon nanoparticles in medicine for cancer therapy. ACS Med. Chem. Lett. 2013, 4, 1012–1013. [Google Scholar] [CrossRef] [PubMed]
- Sponchia, G.; Rizzolio, F.; Hadla, M.; Del Tedesco, A.; Russo, S.C.; Toffoli, G.; Riello, P.; Benedetti, A. Biocompatible tailored zirconia mesoporous nanoparticles with high surface area for theranostic applications. J. Mater. Chem. B 2015, 3, 7300–7306. [Google Scholar] [CrossRef]
- Leonetti, J.P.; Degols, G.; Lebleu, B. Biological activity of oligonucleotide-poly(l-lysine) conjugates: Mechanism of cell uptake. Bioconjug. Chem. 1990, 1, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Posocco, B.; Dreussi, E.; de Santa, J.; Toffoli, G.; Abrami, M.; Musiani, F.; Grassi, M.; Farra, R.; Tonon, F.; Grassi, G.; et al. Polysaccharides for the Delivery of Antitumor Drugs. Materials 2015, 8, 2569–2615. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, T.; Aljaeid, B. Preparation characterization and potential application of chitosan, chitosan derivates, and chitosan metal nanoparticles in pharmaceutical drug delivery. Drug Des. Dev. Ther. 2016, 10, 483–507. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wang, J. Delivery systems for siRNA drug development in cancer therapy. Asian J. Pharm. Sci. 2015, 10, 1–12. [Google Scholar] [CrossRef]
- Hobel, S.; Aigner, A. Polyethylenimines for siRNA and miRNA delivery in vivo. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2013, 5, 484–501. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zheng, M.; Librizzi, D.; Renette, T.; Merkel, O.M.; Kissel, T. Efficient and Tumor Targeted siRNA Delivery by Polyethylenimine-graft-polycaprolactone-block-poly(ethylene glycol)-folate (PEI-PCL-PEG-Fol). Mol. Pharm. 2016, 13, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.J.; Bentley, M.D.; Harris, J.M. Chemistry for peptide and protein PEGylation. Adv. Drug Deliv. Rev. 2002, 54, 459–476. [Google Scholar] [CrossRef]
- Bao, Y.; Jin, Y.; Chivukula, P.; Zhang, J.; Liu, Y.; Liu, J.; Clamme, J.P.; Mahato, R.I.; Ng, D.; Ying, W.; et al. Effect of PEGylation on biodistribution and gene silencing of siRNA/lipid nanoparticle complexes. Pharm. Res. 2013, 30, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan, P.; Mallory, E.; Malapit, M.; Hayes, D., Jr.; Mansour, H.M. Inhalable PEGylated Phospholipid Nanocarriers and PEGylated Therapeutics for Respiratory Delivery as Aerosolized Colloidal Dispersions and Dry Powder Inhalers. Pharmaceutics 2014, 6, 333–353. [Google Scholar] [CrossRef] [PubMed]
- Azimi, B.; Nourpanak, P.; Rabiee, M.; Arab, S. Poly(e-caprolactone) Fiber: An Overview. J. Eng. Fibers Fabr. 2014, 9, 74–90. [Google Scholar]
- Mensink, M.A.; Frijlink, H.W.; van der Voort Maarschalk, K.; Hinrichs, W.L. Inulin, a flexible oligosaccharide. II: Review of its pharmaceutical applications. Carbohydr. Polym. 2015, 134, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Sardo, C.; Farra, R.; Licciardi, M.; Dapas, B.; Scialabba, C.; Giammona, G.; Grassi, M.; Grassi, G.; Cavallaro, G. Development of a simple, biocompatible and cost-effective Inulin-Diethylenetriamine based siRNA delivery system. Eur. J. Pharm. Sci. 2015, 75, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Loh, X.J. Cyclodextrin-based supramolecular architectures: Syntheses, structures, and applications for drug and gene delivery. Adv. Drug Deliv. Rev. 2008, 60, 1000–1017. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yuan, S.X.; Zhao, L.H.; Wang, C.; Ni, J.S.; Wang, Z.G.; Lin, C.; Wu, M.C.; Zhou, W.P. Ligand-directed stearic acid grafted chitosan micelles to increase therapeutic efficacy in hepatic cancer. Mol. Pharm. 2015, 12, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Cazejust, J.; Bessoud, B.; Colignon, N.; Garcia-Alba, C.; Planche, O.; Menu, Y. Hepatocellular carcinoma vascularization: From the most common to the lesser known arteries. Diagn. Interv. Imaging 2014, 95, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.F.; Poon, R.T. Vascular changes in hepatocellular carcinoma. Anat. Rec. (Hoboken) 2008, 291, 721–734. [Google Scholar] [CrossRef] [PubMed]
- Piscaglia, F.; Bolondi, L. The intermediate hepatocellular carcinoma stage: Should treatment be expanded? Dig. Liver Dis. 2010, 42 (Suppl. 3), S258–S263. [Google Scholar] [CrossRef]
- Gaba, R.C.; Schwind, R.M.; Ballet, S. Mechanism of Action, Pharmacokinetics, Efficacy, and Safety of Transarterial Therapies Using Ethiodized Oil: Preclinical Review in Liver Cancer Models. J. Vasc. Interv. Radiol. 2018, 29, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.Y.; Xu, L.F.; Wang, W.D.; Huang, Q.S.; Sun, H.L.; Chen, Y.T. Transarterial embolization combined with RNA interference targeting hypoxia-inducible factor-1α for hepatocellular carcinoma: A preliminary study of rat model. J. Cancer Res. Clin. Oncol. 2017, 143, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F.; Nagy, J.A.; Dvorak, J.T.; Dvorak, A.M. Identification and characterization of the blood vessels of solid tumors that are leaky to circulating macromolecules. Am. J. Pathol. 1988, 133, 95–109. [Google Scholar] [PubMed]
- Iyer, A.K.; Khaled, G.; Fang, J.; Maeda, H. Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discov. Today 2006, 11, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, H.; Baluk, P.; Morikawa, S.; McLean, J.W.; Thurston, G.; Roberge, S.; Jain, R.K.; McDonald, D.M. Openings between defective endothelial cells explain tumor vessel leakiness. Am. J. Pathol. 2000, 156, 1363–1380. [Google Scholar] [CrossRef]
- D’Apolito, R.; Tomaiuolo, G.; Taraballi, F.; Minardi, S.; Kirui, D.; Liu, X.; Cevenini, A.; Palomba, R.; Ferrari, M.; Salvatore, F.; et al. Red blood cells affect the margination of microparticles in synthetic microcapillaries and intravital microcirculation as a function of their size and shape. J. Control Release 2015, 217, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Nel, A.E.; Madler, L.; Velegol, D.; Xia, T.; Hoek, E.M.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding biophysicochemical interactions at the nano-bio interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Liu, W.; Misra, P.; Tanaka, E.; Zimmer, J.P.; Itty, I.B.; Bawendi, M.G.; Frangioni, J.V. Renal clearance of quantum dots. Nat. Biotechnol. 2007, 25, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.F.; Yang, T.F.; Huang, C.T.; Chen, M.C.; Sung, H.W. Preparation of nanoparticles composed of poly(gamma-glutamic acid)-poly(lactide) block copolymers and evaluation of their uptake by HepG2 cells. J. Control Release 2005, 105, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, K.M.; MacParland, S.A.; Ma, X.Z.; Spetzler, V.N.; Echeverri, J.; Ouyang, B.; Fadel, S.M.; Sykes, E.A.; Goldaracena, N.; Kaths, J.M.; et al. Mechanism of hard-nanomaterial clearance by the liver. Nat. Mater. 2016, 15, 1212–1221. [Google Scholar] [CrossRef] [PubMed]
- Walkey, C.D.; Olsen, J.B.; Guo, H.; Emili, A.; Chan, W.C. Nanoparticle size and surface chemistry determine serum protein adsorption and macrophage uptake. J. Am. Chem. Soc. 2012, 134, 2139–2147. [Google Scholar] [CrossRef] [PubMed]
- Arnida; Janat-Amsbury, M.M.; Ray, A.; Peterson, C.M.; Ghandehari, H. Geometry and surface characteristics of gold nanoparticles influence their biodistribution and uptake by macrophages. Eur. J. Pharm. Biopharm. 2011, 77, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Ogawara, K.; Furumoto, K.; Nagayama, S.; Minato, K.; Higaki, K.; Kai, T.; Kimura, T. Pre-coating with serum albumin reduces receptor-mediated hepatic disposition of polystyrene nanosphere: Implications for rational design of nanoparticles. J. Control Release 2004, 100, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Camner, P.; Lundborg, M.; Lastbom, L.; Gerde, P.; Gross, N.; Jarstrand, C. Experimental and calculated parameters on particle phagocytosis by alveolar macrophages. J. Appl. Physiol. (1985) 2002, 92, 2608–2616. [Google Scholar] [CrossRef] [PubMed]
- Beyoglu, D.; Imbeaud, S.; Maurhofer, O.; Bioulac-Sage, P.; Zucman-Rossi, J.; Dufour, J.F.; Idle, J.R. Tissue metabolomics of hepatocellular carcinoma: Tumor energy metabolism and the role of transcriptomic classification. Hepatology 2013, 58, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Li, J.; Li, X.; Mu, H.; Zhang, X.; Shi, Y.; Chu, Y.; Wang, A.; Wu, Z.; Sun, K. Magnetically and pH dual responsive dendrosomes for tumor accumulation enhanced folate-targeted hybrid drug delivery. J. Control Release 2016, 232, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Gullino, P.M.; Clark, S.H.; Grantham, F.H. The Interstitia Fluid of solid tumors. Cancer Res. 1964, 24, 780–794. [Google Scholar] [PubMed]
- Aukland, K.; Reed, R.K. Interstitial-lymphatic mechanisms in the control of extracellular fluid volume. Physiol. Rev. 1993, 73, 1–78. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; Baxter, L.T. Mechanisms of heterogeneous distribution of monoclonal antibodies and other macromolecules in tumors: Significance of elevated interstitial pressure. Cancer Res. 1988, 48, 7022–7032. [Google Scholar] [PubMed]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Lammers, T.; Kiessling, F.; Hennink, W.E.; Storm, G. Drug targeting to tumors: Principles, pitfalls and (pre-) clinical progress. J. Control Release 2012, 161, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Li, Y.; Liu, W.; Jin, L.; Jiang, X.; Wang, X.; Ding, Z.; Peng, Y.; Zhou, J.; Fan, J.; et al. The nanomechanical signature of liver cancer tissues and its molecular origin. Nanoscale 2015, 7, 12998–13010. [Google Scholar] [CrossRef] [PubMed]
- Baenziger, J.U.; Maynard, Y. Human hepatic lectin. Physiochemical properties and specificity. J. Biol. Chem. 1980, 255, 4607–4613. [Google Scholar] [PubMed]
- Craparo, E.F.; Sardo, C.; Serio, R.; Zizzo, M.G.; Bondi, M.L.; Giammona, G.; Cavallaro, G. Galactosylated polymeric carriers for liver targeting of sorafenib. Int. J. Pharm. 2014, 466, 172–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, H.; Lin, K.X.; Zhao, H.; Xing, S.; Li, C.; Liu, F.; Lu, H.Z.; Zhang, Z.; Sun, Y.L.; Yan, X.Y.; et al. Identification of biomarkers for hepatocellular carcinoma by semiquantitative immunocytochemistry. World J. Gastroenterol. 2014, 20, 5826–5838. [Google Scholar] [CrossRef] [PubMed]
- Nakatsura, T.; Yoshitake, Y.; Senju, S.; Monji, M.; Komori, H.; Motomura, Y.; Hosaka, S.; Beppu, T.; Ishiko, T.; Kamohara, H.; et al. Glypican-3, overexpressed specifically in human hepatocellular carcinoma, is a novel tumor marker. Biochem. Biophys. Res. Commun. 2003, 306, 16–25. [Google Scholar] [CrossRef]
- Baumhoer, D.; Tornillo, L.; Stadlmann, S.; Roncalli, M.; Diamantis, E.K.; Terracciano, L.M. Glypican 3 expression in human nonneoplastic, preneoplastic, and neoplastic tissues: A tissue microarray analysis of 4,387 tissue samples. Am. J. Clin. Pathol. 2008, 129, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Jing, S.Q.; Trowbridge, I.S. Identification of the intermolecular disulfide bonds of the human transferrin receptor and its lipid-attachment site. EMBO J. 1987, 6, 327–331. [Google Scholar] [PubMed]
- Sutherland, R.; Delia, D.; Schneider, C.; Newman, R.; Kemshead, J.; Greaves, M. Ubiquitous cell-surface glycoprotein on tumor cells is proliferation-associated receptor for transferrin. Proc. Natl. Acad. Sci. USA 1981, 78, 4515–4519. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Capobianco, A.; Cali, A.; Bellora, F.; Alberti, F.; Righi, L.; Sapino, A.; Camaschella, C.; Malavasi, F. Structural, functional, and tissue distribution analysis of human transferrin receptor-2 by murine monoclonal antibodies and a polyclonal antiserum. Blood 2002, 100, 3782–3789. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.C.; Liu, L.; Wang, X.R.; Shuai, W.P.; Hu, Y.; Han, M.; Gao, J.Q. Folate receptor-targeted liposomes loaded with a diacid metabolite of norcantharidin enhance antitumor potency for H22 hepatocellular carcinoma both in vitro and in vivo. Int. J. Nanomed. 2016, 11, 1395–1412. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Chen, H.; Yu, Y.; Song, J.; Song, H.; Su, X.; Li, W.; Tong, X.; Qian, W.; Wang, H.; et al. Inhibition of hepatocellular carcinoma growth using immunoliposomes for co-delivery of adriamycin and ribonucleotide reductase M2 siRNA. Biomaterials 2013, 34, 10084–10098. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, Y.; Wang, W.; Liu, K. Effect of integrin receptor-targeted liposomal paclitaxel for hepatocellular carcinoma targeting and therapy. Oncol. Lett. 2015, 10, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Leiva, A.; Verdejo, H.; Benitez, M.L.; Martinez, A.; Busso, D.; Rigotti, A. Mechanisms regulating hepatic SR-BI expression and their impact on HDL metabolism. Atherosclerosis 2011, 217, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Ozturk, M.; Wilson, B.; Maki, A.; Ozawa, K.; Koizumi, M.; Endo, K.; Strauss, W.; Shouval, D.; Wands, J. In vivo expression of two novel tumor-associated antigens and their use in immunolocalization of human hepatocellular carcinoma. Hepatology 1989, 9, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Scaggiante, B.; Dapas, B.; Farra, R.; Grassi, M.; Pozzato, G.; Giansante, C.; Fiotti, N.; Tamai, E.; Tonon, F.; Grassi, G. Aptamers as targeting delivery devices or anti-cancer drugs for fighting tumors. Curr. Drug Metab. 2013, 14, 565–582. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Yang, L.; Zhao, X.; Zhang, L.; Zhu, H.; Liu, C.; Tan, W. Targeted delivery of chemotherapy agents using a liver cancer-specific aptamer. PLoS ONE 2012, 7, e33434. [Google Scholar] [CrossRef] [PubMed]
- Scaggiante, B.; Dapas, B.; Farra, R.; Tonon, F.; Abrami, M.; Grassi, M.; Musiani, F.; Zanconati, F.; Pozzato, G.; Grassi, G. Translation Elongation. In Translation and Its Regulation in Cancer Biology and Medicine; Parsyan, A., Ed.; Springer: Berlin, Germany, 2014; pp. 241–265. [Google Scholar]
- Lamberti, A.; Caraglia, M.; Longo, O.; Marra, M.; Abbruzzese, A.; Arcari, P. The translation elongation factor 1A in tumorigenesis, signal transduction and apoptosis: Review article. Amino Acids 2004, 26, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Grassi, G.; Scaggiante, B.; Farra, R.; Dapas, B.; Agostini, F.; Baiz, D.; Rosso, N.; Tiribelli, C. The expression levels of the translational factors eEF1A 1/2 correlate with cell growth but not apoptosis in hepatocellular carcinoma cell lines with different differentiation grade. Biochimie 2007, 89, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Qiu, F.N.; Huang, Y.; Chen, D.Y.; Li, F.; Wu, Y.A.; Wu, W.B.; Huang, X.L. Eukaryotic elongation factor-1alpha 2 knockdown inhibits hepatocarcinogenesis by suppressing PI3K/Akt/NF-kappaB signaling. World J. Gastroenterol. 2016, 22, 4226–4237. [Google Scholar] [CrossRef] [PubMed]
- Schlaeger, C.; Longerich, T.; Schiller, C.; Bewerunge, P.; Mehrabi, A.; Toedt, G.; Kleeff, J.; Ehemann, V.; Eils, R.; Lichter, P.; et al. Etiology-dependent molecular mechanisms in human hepatocarcinogenesis. Hepatology 2008, 47, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, R.; Calvisi, D.F.; Neumann, O.; Kolluru, V.; Wesely, J.; Chen, X.; Wang, C.; Wuestefeld, T.; Ladu, S.; Elgohary, N.; et al. EEF1A2 inactivates p53 by way of PI3K/AKT/mTOR-dependent stabilization of MDM4 in hepatocellular carcinoma. Hepatology 2014, 59, 1886–1899. [Google Scholar] [CrossRef] [PubMed]
- Kovesdi, I.; Reichel, R.; Nevins, J.R. Role of an adenovirus E2 promoter binding factor in E1A-mediated coordinate gene control. Proc. Natl. Acad. Sci. USA 1987, 84, 2180–2184. [Google Scholar] [CrossRef] [PubMed]
- Farra, R.; Grassi, G.; Tonon, F.; Abrami, M.; Grassi, M.; Pozzato, G.; Fiotti, N.; Forte, G.; Dapas, B. The Role of the Transcription Factor E2F1 in Hepatocellular Carcinoma. Curr. Drug Deliv. 2017, 14, 272–281. [Google Scholar] [PubMed]
- Conner, E.A.; Lemmer, E.R.; Omori, M.; Wirth, P.J.; Factor, V.M.; Thorgeirsson, S.S. Dual functions of E2F-1 in a transgenic mouse model of liver carcinogenesis. Oncogene 2000, 19, 5054–5062. [Google Scholar] [CrossRef] [PubMed]
- Lukas, E.R.; Bartley, S.M.; Graveel, C.R.; Diaz, Z.M.; Dyson, N.; Harlow, E.; Yamasaki, L.; Farnham, P.J. No effect of loss of E2F1 on liver regeneration or hepatocarcinogenesis in C57BL/6J or C3H/HeJ mice. Mol. Carcinog. 1999, 25, 295–303. [Google Scholar] [CrossRef]
- Satow, R.; Shitashige, M.; Kanai, Y.; Takeshita, F.; Ojima, H.; Jigami, T.; Honda, K.; Kosuge, T.; Ochiya, T.; Hirohashi, S.; et al. Combined functional genome survey of therapeutic targets for hepatocellular carcinoma. Clin. Cancer Res. 2010, 16, 2518–2528. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zhou, Z.; Qiu, N.; Shen, Y. Rational Design of Cancer Nanomedicine: Nanoproperty Integration and Synchronization. Adv. Mater. 2017, 29, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, D.W.; Davis, M.E. Insights into the kinetics of siRNA-mediated gene silencing from live-cell and live-animal bioluminescent imaging. Nucleic Acids Res. 2006, 34, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Tang, C.; Yin, C. Oral delivery of shRNA and siRNA via multifunctional polymeric nanoparticles for synergistic cancer therapy. Biomaterials 2014, 35, 4589–4600. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Chen, J.; Shi, M.; Deivasigamani, A.; Ooi, L.L.; Hui, K.M. The over-expression of survivin enhances the chemotherapeutic efficacy of YM155 in human hepatocellular carcinoma. Oncotarget 2015, 6, 5990–6000. [Google Scholar] [CrossRef] [PubMed]
- Beierle, E.A.; Nagaram, A.; Dai, W.; Iyengar, M.; Chen, M.K. VEGF-mediated survivin expression in neuroblastoma cells. J. Surg. Res. 2005, 127, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.Y.; Kuo, W.T.; Chou, M.J.; Huang, Y.Y. Co-delivery of anti-vascular endothelial growth factor siRNA and doxorubicin by multifunctional polymeric micelle for tumor growth suppression. J. Biomed. Mater. Res. A 2011, 97, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.X.; Xiong, M.H.; Wang, Y.C.; Zhu, J.; Wang, J. N-acetylgalactosamine functionalized mixed micellar nanoparticles for targeted delivery of siRNA to liver. J. Control Release 2013, 166, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, G.; Farra, R.; Craparo, E.F.; Sardo, C.; Porsio, B.; Giammona, G.; Perrone, F.; Grassi, M.; Pozzato, G.; Grassi, G.; et al. Galactosylated polyaspartamide copolymers for siRNA targeted delivery to hepatocellular carcinoma cells. Int. J. Pharm. 2017, 525, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Tang, C.; Yin, C. Effect of binding affinity for siRNA on the in vivo antitumor efficacy of polyplexes. Biomaterials 2013, 34, 5317–5327. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Tang, C.; Yin, C. Enhanced antitumor efficacies of multifunctional nanocomplexes through knocking down the barriers for siRNA delivery. Biomaterials 2015, 44, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Wang, J.; Zhang, L.; Shen, S.; Guo, R.; Yang, Y.; Chen, W.; Wang, Y.; Chen, G.; Shuai, X. Theranostical nanosystem-mediated identification of an oncogene and highly effective therapy in hepatocellular carcinoma. Hepatology 2016, 63, 1240–1255. [Google Scholar] [CrossRef] [PubMed]
- Qu, C.; He, D.; Lu, X.; Dong, L.; Zhu, Y.; Zhao, Q.; Jiang, X.; Chang, P.; Jiang, X.; Wang, L.; et al. Salt-inducible Kinase (SIK1) regulates HCC progression and WNT/beta-catenin activation. J. Hepatol. 2016, 64, 1076–1089. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Wang, C.C.; Choy, K.W.; Du, Q.; Chen, J.; Wang, Q.; Li, L.; Chung, T.K.; Tang, T. Therapeutic potentials of gene silencing by RNA interference: Principles, challenges, and new strategies. Gene 2014, 538, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Gong, F.; Pang, P.; Shen, M.; Zhu, K.; Cheng, D.; Liu, Z.; Shan, H. An RGD-modified MRI-visible polymeric vector for targeted siRNA delivery to hepatocellular carcinoma in nude mice. PLoS ONE 2013, 8, e66416. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Extended Name | Abbreviation | References |
|---|---|---|
| Asialoglycoprotein receptor | ASGP-R | [80,81,82] |
| Glypican-3 | GPC3 | [83,84] |
| Transferrin receptor | TfR | [85,86,87] |
| Folic acid receptor | FR | [88] |
| Epidermal growth factor receptor | EGFR | [89] |
| αvβ3 and αvβ5 integrins | [90] | |
| Scavenger receptor class B type I | SR-BI receptor | [91] |
| Homodimeric glycoprotein | AF-20 antigen | [92] |
| Delivery Material | HCC Targeting Antigen | HCC Model | siRNA mRNA Target | Reference |
|---|---|---|---|---|
| Galactose modified trimethylchitosan-cystein (GTC) | ASGP-R | BEL-7402 | Survivin and VEGF | [108] |
| PEI grafted with stearic acid (PEI-SA) | FR | HuH-7 | VEGF | [111] |
| GalNac- PEG-b-PCL and PCL-b-PPEEA | ASGP-R | Primary hepatocytes | apolipoprotein B | [112] |
| Inulin and diethylentriamine (Inu-DETA) on α,β-poly-(N-2-hydroxyethyl)-d,l-aspartamide (PHEA) and DETA and PEG) and GAL molecules (PHEA-DETA-PEG-GAL) | Trafficking specificity | JHH6 | E2F1 | [52] |
| ASGP-R | JHH6 | E2F1 | [113] |
| Delivery Material and Particle Size | HCC Targeting Antigen | HCC Model | siRNA mRNA Target | Reference |
|---|---|---|---|---|
| Urocanic acid-modified galactosylated trimethyl chitosan (UA-GT) 170 nm | ASGP-R | QGY-7703 and mouse xenograft subcutaneous model (systemic delivery) | VEGF | [115] |
| Galactose modified trymethil chitosan-cystein (GTC) 130–160 nm | ASGP-R | xenograft mice model of HCC (oral administration) | Survivin and VEGF | [108] |
| GTCs polyplexes with distinct siRNA binding affinity 135–170 nm | ASGP-R | QGY-7703 and xenograft mice model of HCC (intra-tumor injection) | VEGF | [114] |
| FA-PEG-g-PEI-SPION 60 nm | FR | orthotropic and xenograft models (systemic delivery) | TBLR1 | [116] |
| RGD-PEG-g-PEI-SPION 122 nm | αvβ3 and αvβ5 integrins | Bel-7402 and mouse xenograft subcutaneous model (systemic delivery) | Survivin | [119] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farra, R.; Musiani, F.; Perrone, F.; Čemažar, M.; Kamenšek, U.; Tonon, F.; Abrami, M.; Ručigaj, A.; Grassi, M.; Pozzato, G.; et al. Polymer-Mediated Delivery of siRNAs to Hepatocellular Carcinoma: Variables Affecting Specificity and Effectiveness. Molecules 2018, 23, 777. https://doi.org/10.3390/molecules23040777
Farra R, Musiani F, Perrone F, Čemažar M, Kamenšek U, Tonon F, Abrami M, Ručigaj A, Grassi M, Pozzato G, et al. Polymer-Mediated Delivery of siRNAs to Hepatocellular Carcinoma: Variables Affecting Specificity and Effectiveness. Molecules. 2018; 23(4):777. https://doi.org/10.3390/molecules23040777
Chicago/Turabian StyleFarra, Rossella, Francesco Musiani, Francesca Perrone, Maja Čemažar, Urška Kamenšek, Federica Tonon, Michela Abrami, Aleš Ručigaj, Mario Grassi, Gabriele Pozzato, and et al. 2018. "Polymer-Mediated Delivery of siRNAs to Hepatocellular Carcinoma: Variables Affecting Specificity and Effectiveness" Molecules 23, no. 4: 777. https://doi.org/10.3390/molecules23040777
APA StyleFarra, R., Musiani, F., Perrone, F., Čemažar, M., Kamenšek, U., Tonon, F., Abrami, M., Ručigaj, A., Grassi, M., Pozzato, G., Bonazza, D., Zanconati, F., Forte, G., El Boustani, M., Scarabel, L., Garziera, M., Russo Spena, C., De Stefano, L., Salis, B., ... Dapas, B. (2018). Polymer-Mediated Delivery of siRNAs to Hepatocellular Carcinoma: Variables Affecting Specificity and Effectiveness. Molecules, 23(4), 777. https://doi.org/10.3390/molecules23040777