Chiral Chalcogen Bond Donors Based on the 4,4′-Bipyridine Scaffold
by
 , , ,
, , ,
Robin Weiss
1, 
Emmanuel Aubert
2 ,
,
Paola Peluso
3,* ,
,
Sergio Cossu
4,
Patrick Pale
1 and 
Victor Mamane
1,* 1
Strasbourg Institut of Chemistry, UMR CNRS 7177, Team LASYROC, 1 rue Blaise Pascal, University of Strasbourg, 67008 Strasbourg CEDEX, France
2
Crystallography, Magnetic Resonance and Modelisations (CRM2), UMR CNRS 7036, University of Lorraine, Bd des Aiguillettes, 54506 Vandoeuvre-les-Nancy, France
3
Institute of Biomolecular Chemistry ICB, CNR, Secondary branch of Sassari, Traversa La Crucca 3, Regione Baldinca, 07100 Li Punti, Sassari, Italy
4
Department of Molecular Science and Nanosystems DSMN, Ca’ Foscari University of Venice, Via Torino 155, 30172 Mestre Venice, Italy
*
Authors to whom correspondence should be addressed.
Molecules 2019, 24(24), 4484; https://doi.org/10.3390/molecules24244484
Submission received: 7 November 2019
/
Revised: 29 November 2019
/
Accepted: 3 December 2019
/
Published: 6 December 2019
(This article belongs to the Special Issue Bipyridines: Synthesis, Functionalization and Applications)
Abstract
:Organocatalysis through chalcogen bonding (ChB) is in its infancy, as its proof-of-principle was only reported in 2016. Herein, we report the design and synthesis of new chiral ChB donors, as well as the catalytic activity evaluation of the 5,5′-dibromo-2,2′-dichloro-3-((perfluorophenyl)selanyl)-4,4′-bipyridine as organocatalyst. The latter is based on the use of two electron-withdrawing groups, a pentafluorophenyl ring and a tetrahalo-4,4′-bipyridine skeleton, as substituents at the selenium center. Atropisomery of the tetrahalo-4,4′-bipyridine motif provides a chiral environment to these new ChB donors. Their synthesis was achieved through either selective lithium exchange and trapping or a site-selective copper-mediated reaction. Pure enantiomers of the 3-selanyl-4,4′-bipyridine were obtained by high performance liquid chromatography enantioseparation on specific chiral stationary phase, and their absolute configuration was assigned by comparison of the measured and calculated electronic circular dichroism spectra. The capability of the selenium compound to participate in σ-hole-based interactions in solution was studied by 19F NMR. Even if no asymmetric induction has been observed so far, the new selenium motif proved to be catalytically active in the reduction of 2-phenylquinoline by Hantzsch ester.
1. Introduction
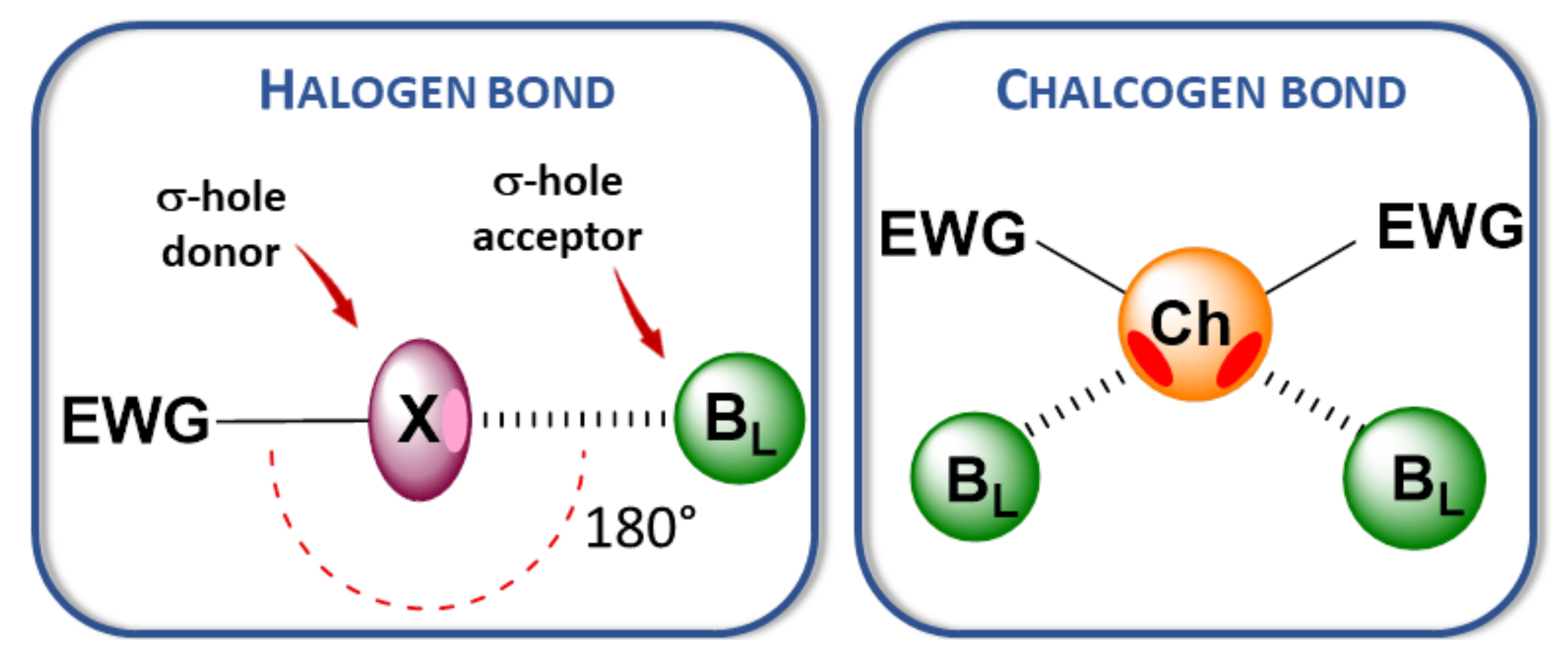
The term chalcogen bond (ChB) describes the intermolecular interaction occurring between a Lewis base (BL) and a group 16 atom (S, Se, Te) which behaves like Lewis acid, according to the recommendations of the International Union of Pure and Applied Chemistry (IUPAC) (net attractive interaction between an electrophilic region associated with a chalcogen atom in a molecular entity and a nucleophilic region in another, or the same, molecular entity) [1,2,3]. ChB belongs to the family of σ-hole-based interactions [4], among which the most prominent and most studied is halogen bonding (XB) [5,6,7]. ChB possesses many similarities with XB [8,9], but with a different interaction pattern, because up to two σ-hole···BL axes can be identified on a Ch atom due to its divalent nature (Figure 1). Although noncovalent contacts involving sulfur have been recognized in biological molecules during the past decade [10], it is only recently that a few applications involving ChB in solid state [11] and in solution [12] have appeared, in particular those involving heavier elements such as selenium and tellurium.
Among the applications based on σ-hole interactions in solution, catalysis represents an active research area. While several catalysts acting as XB donor have been described during the last decade [6,13,14], the first examples of ChB-based catalysis have been very recently reported by the groups of Matile [15,16,17,18], Huber [19,20,21], and Wang [22]. Other recent applications of ChB concern anion binding [23,24] and transport [25,26,27].
Introduction of chirality in systems able to interact through noncovalent bonds can have a significant impact in many areas, including biology, molecular recognition, catalysis, and material sciences [28,29]. Unlike XB-based processes, for which a number of chiral donor molecules were developed in the last few years [5,6,12,30], only very few applications using chiral ChB donors have been reported. Beer’s group described a chiral selenium-containing receptor for enantiomeric discrimination of dicarboxylates [24]. Following previous studies on XB-based high performance liquid chromatography (HPLC) enantioseparations [31,32,33,34,35,36], our groups demonstrated that ChB can participate in the recognition process underlying the enantioseparation of sulfur-containing atropisomeric 4,4′-bipyridine 1 on a cellulose-based chiral stationary phase [37].
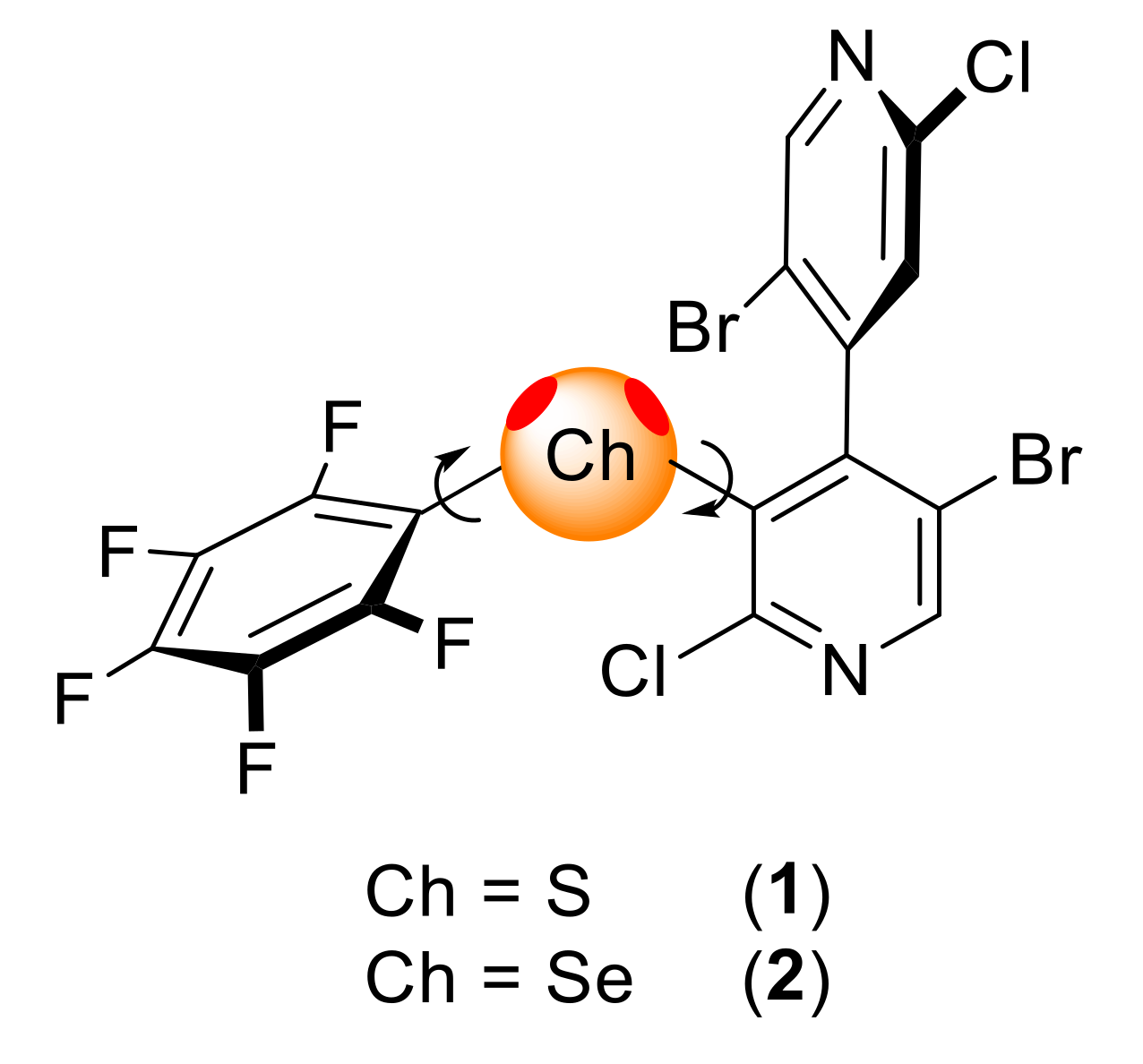
On this basis, we report here our results on the synthesis of thio- and seleno-substituted 4,4′-bipyridines as ChB donors and the evaluation of compounds 1 and 2 as organocatalysts in the reduction of 2-phenylquinoline (Figure 2). For this purpose, an improved synthetic protocol for the synthesis of compound 1 was developed and applied to the preparation of derivative 2. Compounds 1 and 2 were active as ChB donor catalysts in the reduction of 2-phenylquinoline, but without enantioselectivity induction. Moreover, studying the behavior of 1 and 2 in the presence of chloride ions by 19F NMR revealed that these compounds form σ-hole bonds in solution.
2. Results
2.1. Synthesis
The thio-substituted 4,4′-bipyridine 1 was prepared in 21% yield from the readily available pentahalogeno-4,4′-bipyridine 3 [38] by selective iodine/lithium exchange and trapping with bis(pentafluorophenyl)disulfide 4, according to our own method [37]. The same conditions were used to prepare the selenium derivatives 2, 6, and 7 starting from 3 and the corresponding diselenides 5a–c (Scheme 1). Although the expected products could be observed by NMR in the reaction mixtures, their purification proved to be very challenging, and the final products could only be isolated with, respectively, 34, 20, and 51% yield through HPLC purification.
These difficulties and the resulting modest yields led us to consider the use of a cross-coupling reaction involving 4,4′-bipyridine 3 as starting material. Indeed, we already succeeded in selective Pd-catalyzed couplings on halogenated 4,4′-bipyridines [38,39]. However, the introduction of SC6F5 and SeC6F5 groups onto haloaromatics through a cross-coupling reaction is poorly described in the literature. Yagupolskii and coworkers described their copper-mediated coupling with several iodoaromatics [40], and later, Haupt and coworkers applied this methodology to the polyfunctionalization of corannulene [41]. On these bases, we screened several conditions starting from 3 (Table 1), focusing on selectivity, which obviously is a major issue considering the number of potentially reactive carbon–halogen positions.
Methodology optimization was performed by using disulfide 4 (entries 1–10) in order to find the best conditions for the selective mono-functionalization. The reactive copper complex [CuSC6F5] was in situ produced by mixing 4 with two equivalents of copper as a powder in N-methyl-2-pyrrolidone (NMP) at room temperature for 3 h. Bipyridine 3 was then added and the mixture was heated for 16 h at a given temperature. We first evaluated the effect of the temperature on the reactivity and selectivity starting with 1.3 eq. of 4 (entries 1–3). At 80 °C, the reaction was not complete and a substantial amount of starting product was still present (entry 1). At 100 °C, the conversion was higher but the over-coupling compound 8 appeared due to the insertion of a second SC6F5 group on the more substituted and electron-poor pyridine ring (see Supplementary Information for NMR details). Under these conditions, the expected thiobipyridine 1 could, nevertheless, be isolated in good yield (74%, entry 2). Increasing further the temperature (130 °C) induced more formation of 8, allowing its isolation with 39% yield (entry 3). While keeping the temperature at 100 °C, the amount of 4 was increased from 0.5 to 4.0 eq. (entries 4–8) in order to evaluate the effect of electrophile concentration. With half equivalent of 4 (i.e., 1 eq. of the active copper species), the conversion was very low (entry 4). Slight improvement was obtained by increasing the amount of 4 to 0.8 eq. (entry 5) and then to 1.0 eq. (entry 6). With 2.0 and 4.0 eq. (entries 7 and 8), the starting bipyridine 3 was completely consumed, but the amount of 8 increased with the increasing amount of copper reagent. No improvement was observed by increasing the temperature to 130 °C while reducing the amount of 4 to 0.5 equiv (entry 9). Rewardingly, with 2.0 eq., the crude reaction mixture was clean and 1 could thus be isolated with 67% yield (entry 7). Mixing all the components without preformation of [CuSC6F5] led to a higher formation of 8 (entry 10). Under conditions optimized for mono-coupling (as in entry 2), the selenobipyridine 2 was obtained with 81% yield, starting from diselenide 5a (entry 11). The bis-coupling product of selenium, analog to 8, was not observed at 100 °C and could not be obtained at 130 °C because decomposition mostly occurred.
2.2. Analyses
2.2.1. HPLC Enantioseparation and Electronic Circular Dichroism (ECD)
The atropisomeric pairs of bipyridines 1, 2, 6, and 7 were obtained in good amounts by means of HPLC multimilligram enantioseparation under normal phase elution conditions. As we previously reported [37], the enantiomers of compound 1 (>99% ee) were recovered by using Chiralpak IA (immobilized amylose tris(3,5-dimethylphenylcarbamate)) as chiral column. On the contrary, the enantioseparations of compounds 2, 6, and 7 were performed on Chiralpak IC (immobilized cellulose tris(3,5-dichlorophenylcarbamate)) (Table 2).
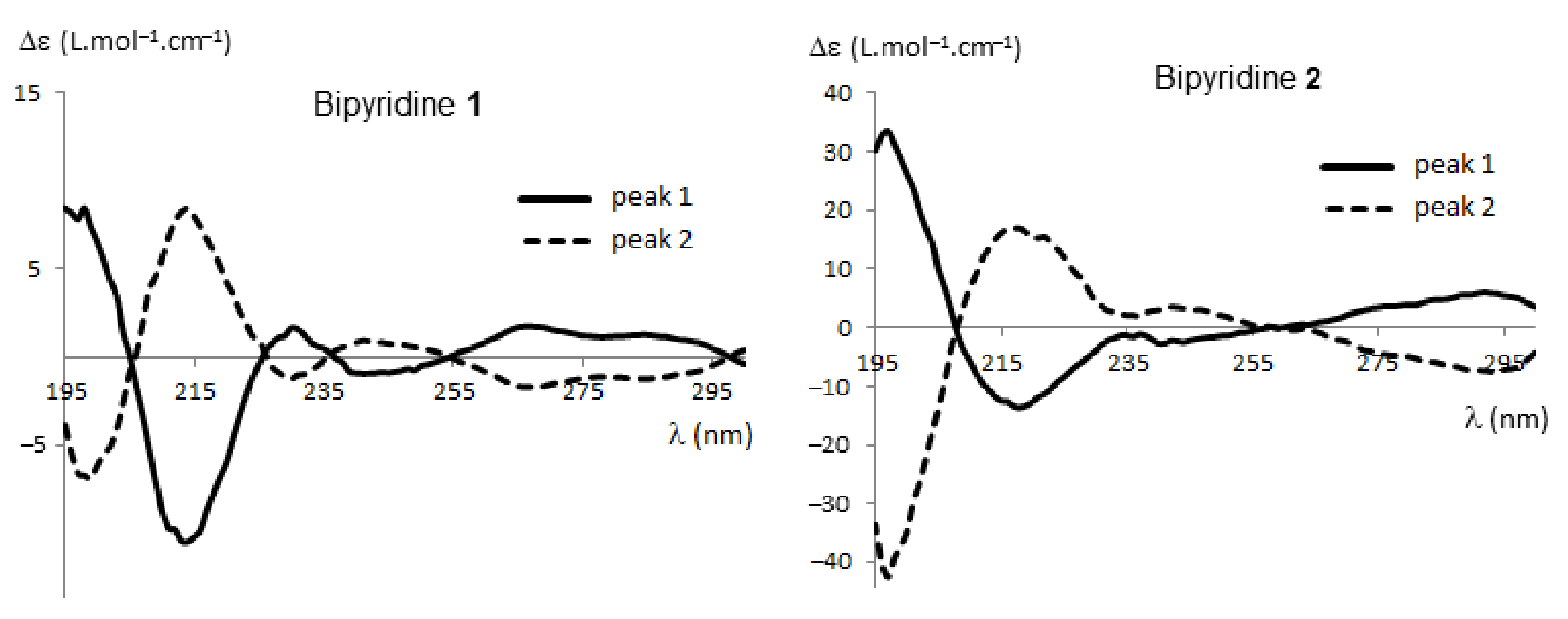
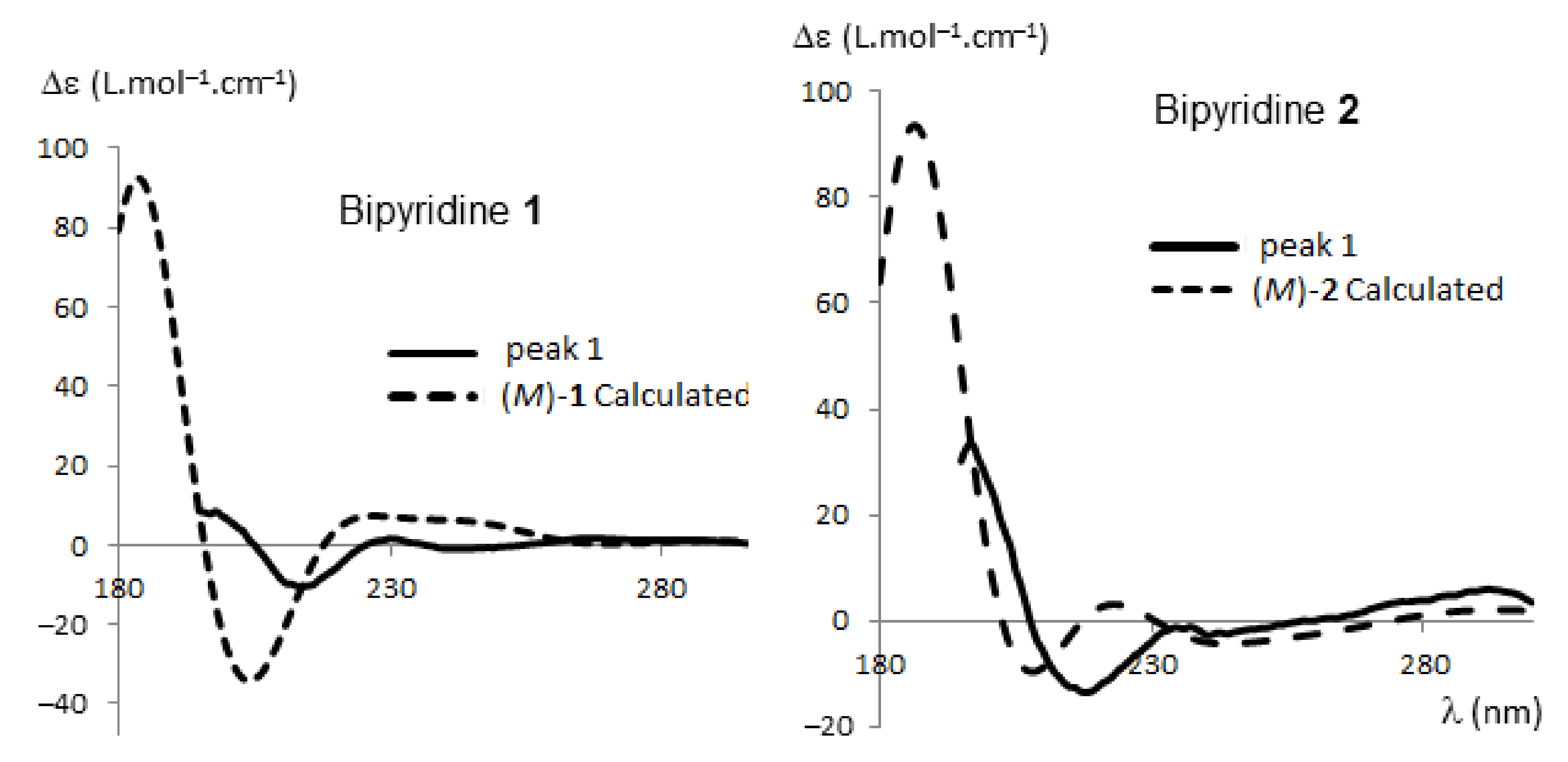
As we demonstrated, the absolute configurations of HPLC-separated enantiomers of polyhalogenated 4,4′-bipyridines can be successfully determined by electronic circular dichroism (ECD) coupled with time-dependent density functional theory (TD-DFT) calculations [37,38,39,42]. These techniques were therefore applied to each enantiomer of 1, 2, 6, and 7. Recorded in ethanol, their ECD spectra showed the expected opposite traces for the two atropisomers (Figure 3; for ECD spectra of 6 and 7, see Figures S1 and S2 in Supplementary Information).
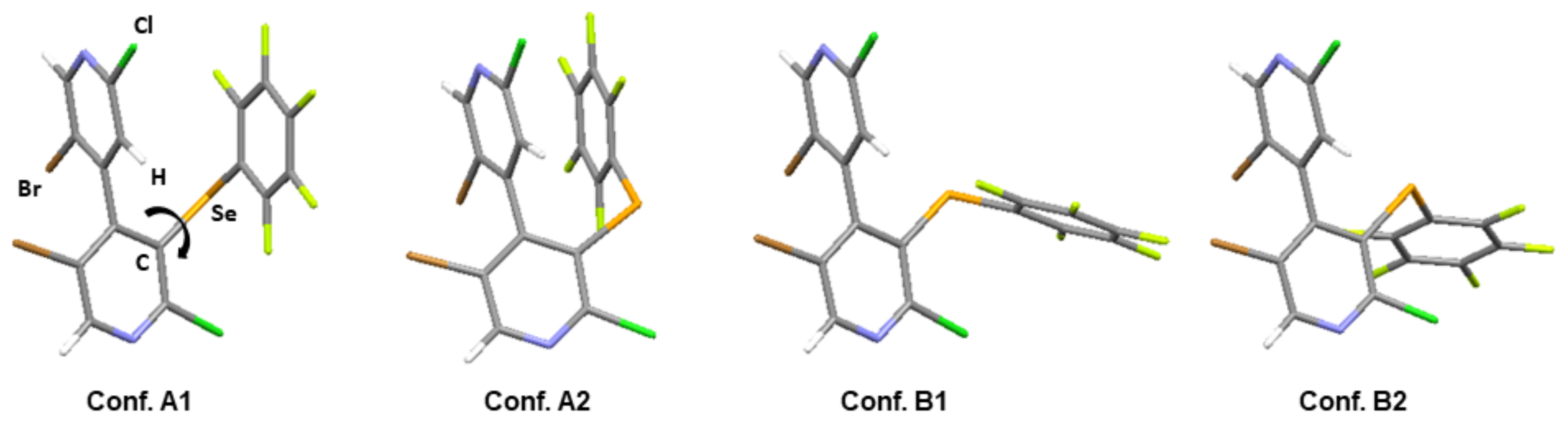
DFT calculations were run at the CAM-B3LYP-D3/6-311+G(d,p) level of theory to explore the conformation distribution in bipyridines 1 and 2 (Figure 4; for details concerning 6 and 7, see Figures S3 and S4 in Supplementary Information) focusing on the pentafluorophenyl group orientation. For compounds 1, 2, and 6, four stable conformations were found. They originate from the relative orientation of the pentaflurophenyl ring that can be in front (conformation A) of the upper pyridine ring or away from it (conformation B) due to rotation around the C–Ch bond. For each of the two conformations, two additional conformers are generated by the relative position of the pentafluorophenyl ring, which can be close to the H (conformations A1 and B1) or to the Br (conformations A2 and B2) of the upper pyridine ring.
The relative energies of all conformers were calculated in EtOH using a polarizable continuum model (PCM), allowing for the determination of their relative population (Table 3). The zero value of energy was ascribed to the more stable conformers, that is, Conf. A2 for 1 and Conf. A1 for 2. We can notice that the distribution of conformers A1, A2, and B1 is different for the two bipyridines, whereas Conf. B2 is negligible in both cases.
The relative population of each conformer, calculated as Boltzmann statistics at 298.15 K, was taken into account in order to calculate the ECD spectra of (M) enantiomers of bipyridines 1, 2, 6, and 7. Comparison of calculated and experimental ECD spectra allowed determining the absolute configurations of the enantiomers (Figure 5; for compounds 6, 7, and complete details, see Figures S9–S14 in Supplementary Information). The agreement between experimental ECD of the first eluted enantiomer and theoretical spectra of (M)-1 was good, taking into account the systematic shift of the calculated spectra to shorter wavelengths (10–15 nm), as evidenced in previous studies using the same DFT function [39]. In conclusion, for all bipyridines 1, 2, 6 and 7, peak 1 corresponds to the (M) enantiomer and peak 2 to the (P) enantiomer. It is worth noting that the (P) configuration for peak 2 of compound 6 was confirmed by X-Ray analysis (see Figure S15 in Supplementary Information).
2.2.2. X-Ray Diffraction
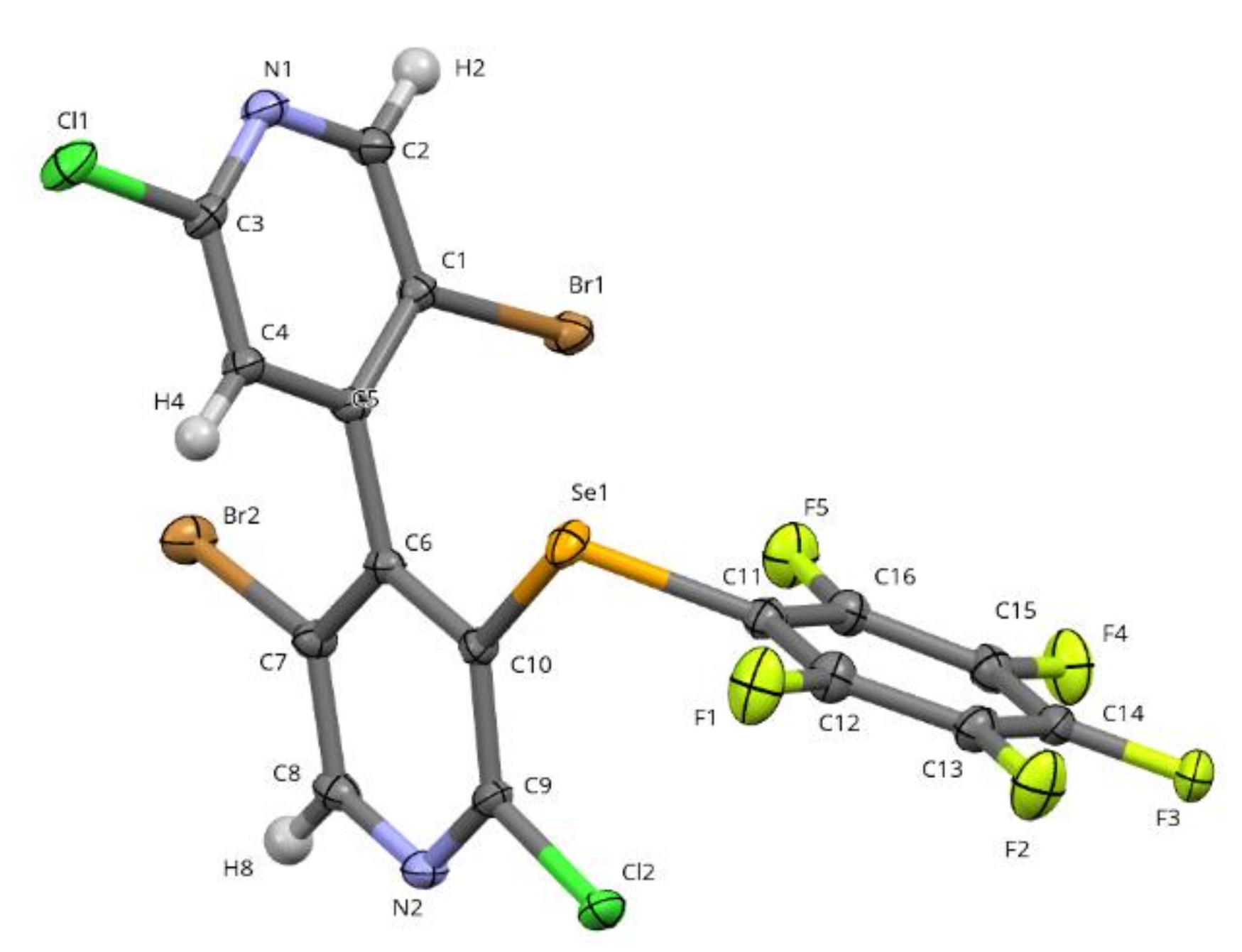
Compounds 1 and 2 are oily, but after some days in the refrigerator (4 °C), some crystals of racemic 2 could be observed and collected. The X-Ray analysis revealed a single molecule in the asymmetric unit, with a conformation very close to the conformer B2 calculated by DFT (root mean square deviation = 0.16Å) (Figure 6).
The structure of 2 displays various intermolecular interactions. Indeed, molecules are interacting about the inversion center through π···π stacking (centroid–centroid distance of 3.7611(9) Å and slippage of 1.293 Å) and cyclic C4–H4···N2 hydrogen bond (H4···N2 = 2.74Å; C4–H4···N2 = 156°) (Figure S16 in Supplementary Information). A type II halogen···halogen bond is observed where the σ-hole of Cl2(1 − x,y,z) is pointing toward the negative crown of Cl1(x,y,z) (Cl2···Cl1 = 3.349Å; C9–Cl2···Cl1 = 177.14°; Cl2···Cl1–C3 = 101.13°) (Figure S17 in Supplementary Information). The π-hole of the pentafluoro ring is in interaction with the electron rich zone of atoms from three neighboring molecules (N2 = (x,1/2 − y,1/2 + z), N1 = (1 + x,y,z) and the negative crown of Br1 = (1 − x,1 − y,1 − z)) (Figure S18 in Supplementary Information).
2.2.3. 19F NMR Spectroscopic Titration with Tetrabutylammonium Chloride (TBACl)
With the aim to probe ChB, binding strength and pattern of bipyridines 1 and 2 with chloride ions were studied in CD2Cl2 by 19F NMR. In addition, to give access to the corresponding binding constants (Ka), this method is expected to give information on the possible competition between the sulfur/selenium σ-holes and the C6F5 π-hole as electrophilic sites toward chloride
The structures of 1 and 2 contain several atoms (Cl, Br, S/Se) or a group of atoms (C6F5) able to interact with chloride ions through their σ- or π-hole. Assuming a preferential interaction of S/Se over Cl/Br with σ-hole acceptors [37], we applied the method of Jin and co-workers [43] in order to identify the major interaction (S/Se σ-hole vs C6F5 π-hole) in compounds 1 and 2. These authors investigated the σ-hole···Cl− (XB) vs. π-hole···Cl− competition by 19F NMR titration of C6F5X (X = F, Cl, Br, I) in the presence of chloride anion. They observed a direct correlation between Δδ of C6F5 upon addition of Cl− and the site of interaction. Thus, with C6F6 and ClC6F5, which are prone to form π-hole interaction with Cl−, the fluorine chemical shifts (δF) moved to lower field. In contrast, with BrC6F5 and IC6F5, which preferentially form a XB, δF moved to higher field.
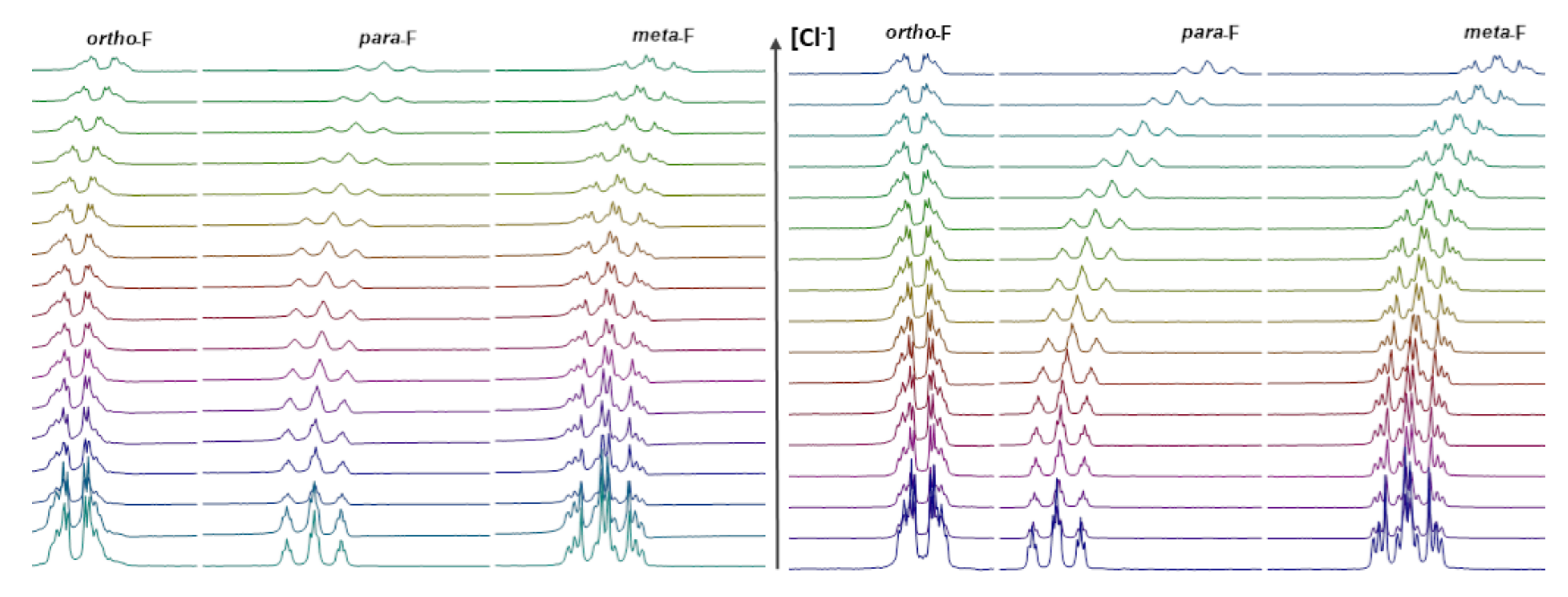
The changes in the 19F NMR chemical shift (Δδ) of the ChC6F5 group were measured by incremental addition of TBACl (from 1 to 200 eq.; see Supplementary Information for details) to a 0.006 mol.L−1 solution of 1 and 2 in CD2Cl2. As shown in Figure 7, δF of C6F5 (para-F) in 1 and 2 move to a higher field with the increase in Cl−. The variation trends of ortho- and meta-F were lower in both compounds, as previously reported [43]. These results show that both bipyridines 1 and 2 interact preferentially with Cl− through the σ-hole of S and Se, respectively.
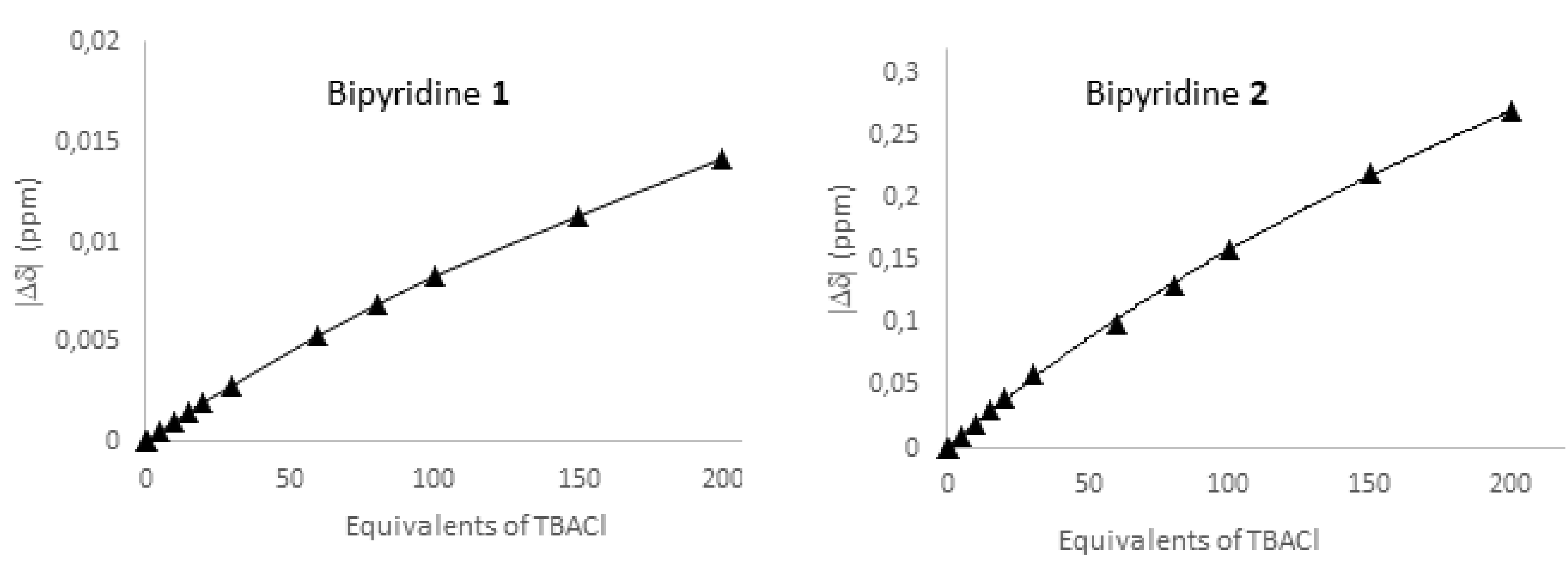
For the calculations of Ka, the measured shifts were plotted against the guest equivalents and the resulting curves were fitted using the website calculator http://supramolecular.org/ (Figure 8 and Supplementary Information for details) [44]. By assuming a 1:1 binding model, Ka values of 0.4 M−1 and 38 M−1 were obtained for compounds 1 and 2, respectively. It is worth noting that the Ka value for 2 (38 M−1) is similar to the one obtained by Matile and coworkers with bis(pentafluorophenyl)selenium [17].
2.3. Catalysis
The hydrogen-transfer reaction using Hantzsch ester (HE) as hydrogen source is a mild method to reduce quinolines and other C=N substrates [45]. Brønsted acids are known to be particularly good catalysts to promote this reaction [46]. In 2008, Bolm showed that haloperfluoroalkanes can be used as XB catalysts for the reduction of 2-phenylquinoline 10 using HE [47]. Since then, this reduction has often been used as a benchmark reaction to evaluate the activity of XB [48,49] and ChB [15,16] catalysts.
We therefore selected the reduction of 2-phenylquinoline 10 as a model substrate for investigating the catalytic ability of the ChB donors 1 and 2 (Table 4). The reaction was followed by 1H NMR over time and it was stopped after 72 h, even if complete conversion was not reached. The resulting tetrahydroquinoline 11 was only isolated and the yield given, when the reaction was completed.
Without catalyst, no conversion was observed after 72 h (entry 6). Rewardingly, with 20 mol% of 1 or 2 as catalysts, the reaction was complete after 48 h, delivering the fully reduced compound 11 with 87% and 94% yields, respectively (entries 1 and 2). Interestingly, with only 5 mol% of 1, a good conversion of 81% was still obtained after 72 h of reaction (entry 3). Under the same conditions, catalyst 2 was even more active, giving full conversion after 72 h, and compound 11 was obtained with an isolated yield of 88% (entry 4). It is interesting to note that with 2, the reaction reached 54% conversion only after 18 h (entry 5).
In the presence of 1 eq. of TBACl, the conversion was lower but not null (entry 7), showing that probably the σ-hole of bromine atoms and/or the π-hole of the C6F5 group took the relay in the catalytic activation of the quinoline substrate. In order to verify this hypothesis, compounds 12 [38] and 13 [37] were tested as catalysts in the reaction (entries 8 and 9). Even with 20 mol%, the reaction did not reach completion after 72 h, showing that the Se atom in compound 2 is the primary site of interaction with the substrate. However, the 62% conversion obtained with 12 is an indication that bromine atoms alone can activate the substrate (entry 8). The presence of the C6F5 group in 13 compared to 12 slightly increased the conversion from 62 to 78%, a result which revealed the modest contribution of the π-hole in the substrate activation (entry 9). With the iodinated compound 3, complete conversion was reached only after 72 h, showing the beneficial effect of the SeC6F5 group compared to iodine atom (entries 10 and 11). Finally, the (P) enantiomer of 2 was employed as catalyst, but unfortunately, no asymmetric induction was observed (entry 12).
3. Discussion
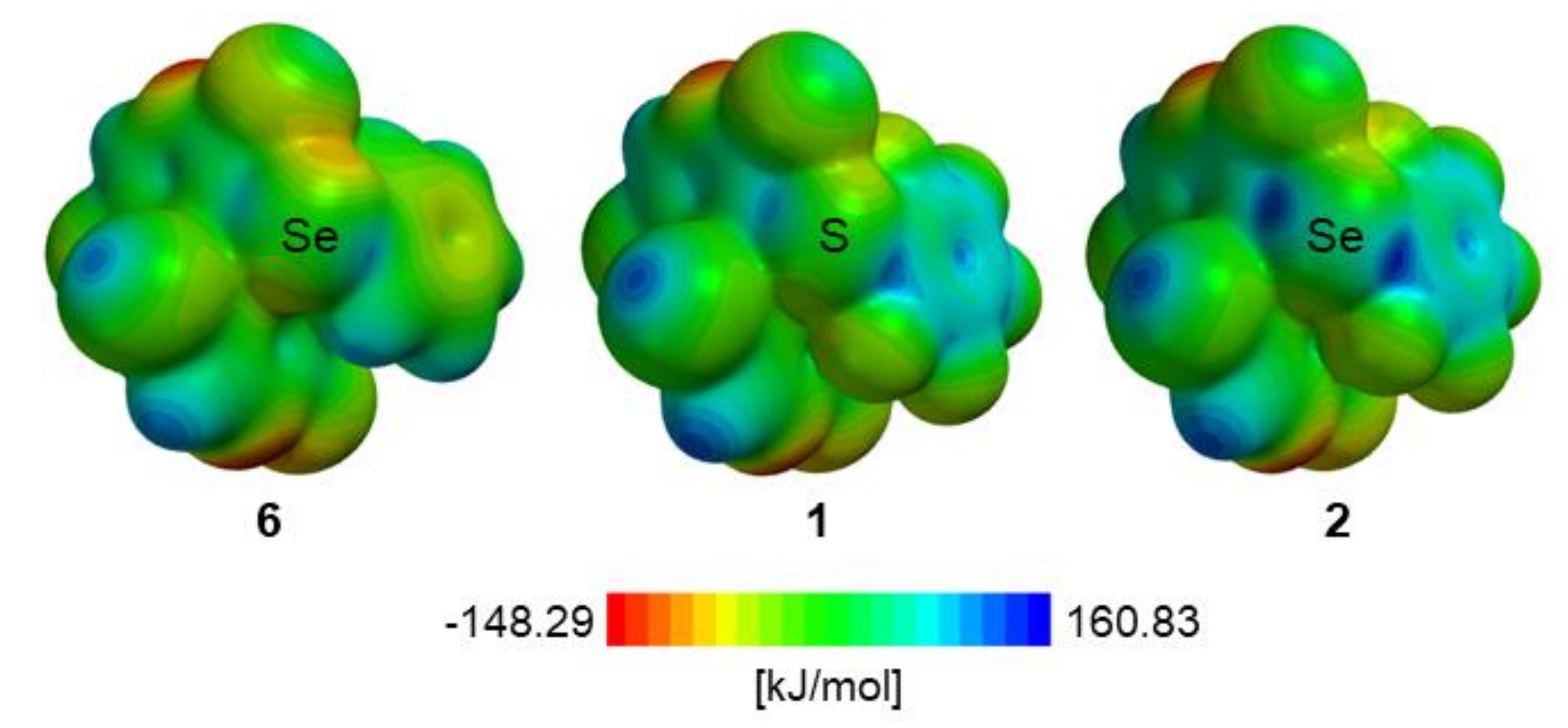
The new motif, which 1 and 2 are based on, was designed by introducing on the Ch site two electron withdrawing groups (EWGs), namely, a pentafluorophenyl ring and a tetrahalo-4,4′-bipyridine scaffold. Indeed, computation of electrostatic potential (EP) maxima on the Ch site indicates that fluorination increase σ-hole depth [50]. In our study, the ChB donor ability ranking for conformationally related (Figure 4, A1-type) structures of 5,5′-dibromo-2,2′-dichloro-3-chalcogeno-4,4′-bipyridines 6, 1, and 2 was determined by evaluating the EP maxima on calculated EP surfaces (DFT/B3LYP/6-311G*), proving that EP maxima values on the Ch σ-hole increase upon fluorination (max EPCh: SePh (6) < SC6F5 (1) < SeC6F5 (2)) (Figure 9 and Table 5). As expected, σ-hole depth increases with the polarizability of the Ch atom. Moreover, the pentafluorophenyl moiety is a well-known π-hole center [51]. On the other hand, the tetrahalo-4,4′-bipyridine skeleton contributes to increase the electrophilic character of the Ch atom and makes the new motif chiral, due to its atropisomeric geometry. In contrast, most reported ChB donors are characterized by a charged heterocyclic structure, which provides sufficient polarization to the Ch atom [2,3,14]. Another peculiarity of the new motif described here is the possibility of rotation around the C–Ch bond, which offers conformational adjustments/tuning during catalysis. It is worth noticing that establishing strong ChBs requires angles R-Ch∙∙∙BL close to 180°, and thus, conformational flexibility of the ChB donor catalyst should increase its catalytic efficiency. In this regard, it is worth noting that, in the compounds so far studied as organocatalysts, the Ch sites are located in a rigid heteroaromatic structure [15,16,17,18,19,20,21].
On this basis, sulfur- and selenium-based chiral catalysts 1 and 2 were prepared by two different methods from chiral 5,5′-dibromo-2,2′-dichloro-3-iodo-4,4′-bipyridine 3. The first method, based on a iodine–lithium exchange followed by quenching with bis(pentafluorophenyl)dichalcogenide, delivered the expected products with low yields. Higher yields were obtained by a copper-mediated coupling of pentafluorophenylthiolate and selenolate with 3. This method proved to be very efficient and selective for the iodinated position. It is therefore expected to proceed with comparable efficiency with other chiral polyhalogenated 4,4′-bipyridines [42] and could represent a new entry to various chiral chalcogen compounds.
The enantiomers of 1 and 2 were separated by HPLC on chiral stationary phase and their absolute configuration was assigned by comparison of the measured and calculated ECD spectra. ECD analysis showed that 5,5′-dibromo-2,2′-dichloro-3-(perfluorophenyl)chalcogeno- 4,4′-bipyridines are present in solution as a mixture of four conformers because of the rotation of the pentafluorophenyl group along the C–Ch bond. Unfortunately, our efforts to detect these conformers in solution by variable temperature 1H NMR were unsuccessful. We could, however, isolate few crystals of the selenium derivative 2 (as a racemate) at low temperature and their XR structure only showed the presence of the minor conformer B2, as revealed from the calculations in solution (Figure 4).
The ability of the chalcogen compounds to drive σ-hole-based interactions in solution was studied through 19F NMR by incremental addition of chloride ions serving as strong acceptors. We observed that δF of ChC6F5 move to a higher field with increasing chloride concentration, showing that both bipyridines interact preferentially with Cl− through the σ-hole of S and Se. Moreover, this study allowed obtaining the binding constants values (Ka) of the 1:1 adducts between compounds 1 or 2 and chloride ion.
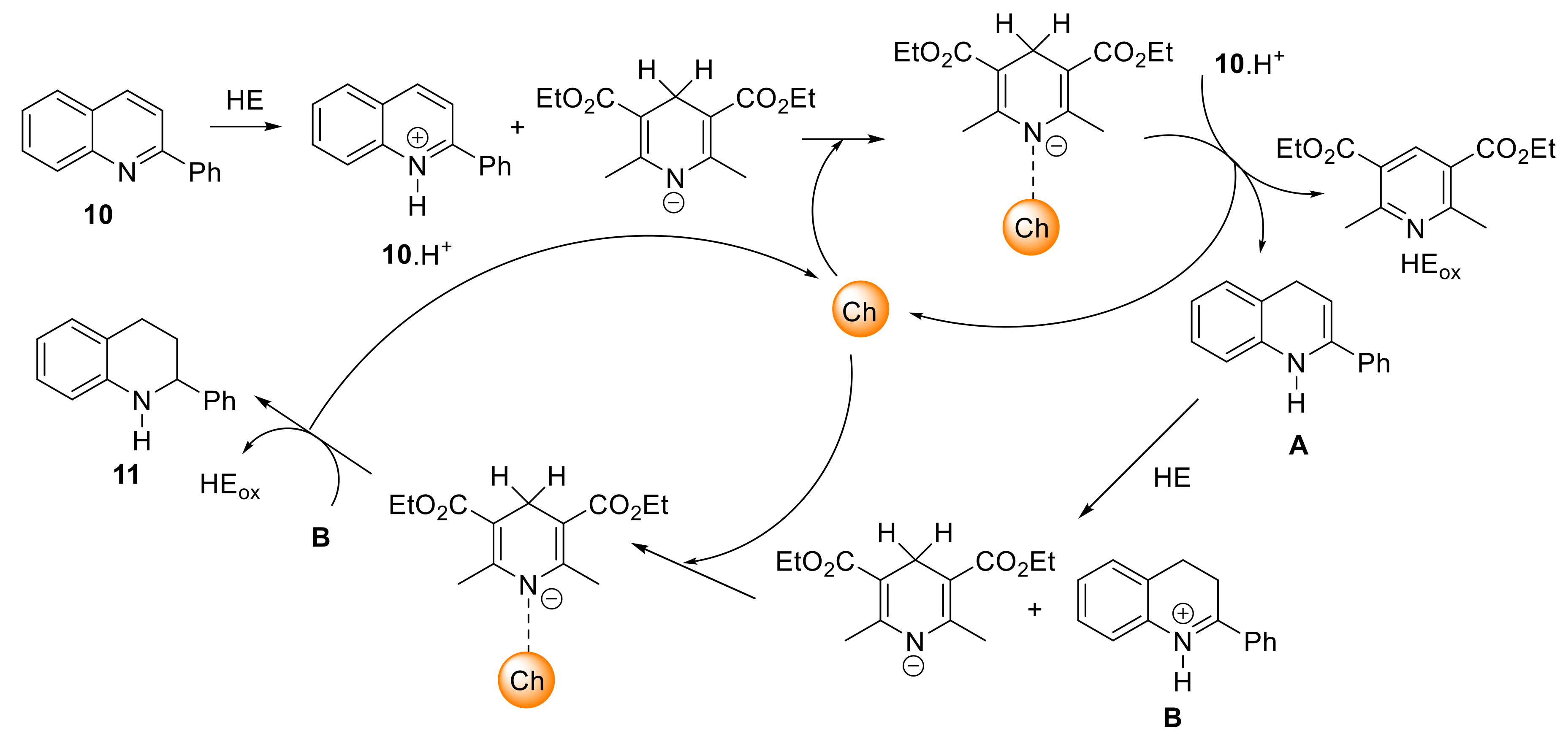
After having shown that derivatives 1 and 2 can behave as ChB donors in solution, their catalytic activity was evaluated in the reduction of 2-phenylquinoline 10. As expected from the Ka values, the Se compound 2 was more active than its S analog 1. Therefore, the study was mainly focused on the use of 2 in comparison with reference compounds where the SeC6F5 group was replaced by CH3 (12), CH2C6F5 (13), and I (3). The reference compound 12 without a σ- or π-hole donor in 3-position was chosen so as to keep the torsional angle between the two pyridine rings similar to compound 2 in order to conserve the same geometry and to analyze only the effect due to the presence of the SeC6F5 group. In agreement with the study in solution, the catalysis results show that 2 interacts in priority through the Se atom with the substrate. It is worth noting that other sites can interact with the substrate, although to a lesser extent: The two bromine atoms in 5,5′-position and the C6F5 group. The last aspect concerns the absence of enantioselectivity when using enantiomerically pure (P)-2. This result may be due to the complex mechanism of the selected reaction (Scheme 2).
After protonation of quinoline 10 by Hantzsch ester (HE), the first reduction occurs to give enamine intermediate A. The latter is protonated to B and then further reduced by the second equivalent of HE. Both reductions can be activated by the ChB donor catalyst. A recent DFT study by Wang et al. with a XB donor catalyst showed that in both reductions, the halogen atom interacts with the deprotonated HE, thus facilitating the hydride transfer [52]. If the same activation mode occurs with catalyst (P)-2 through ChB, the chiral bipyridine backbone would be placed far from the C=N bond of the substrate, and such spatial arrangement could explain the lack of asymmetric induction.
On these bases, we are currently working on the synthesis of analogs of 2, where bromines are exchanged by chlorines or fluorines and the C6F5 group by a CF3. Furthermore, the search for new ChB-catalyzed reactions, for which asymmetric induction could be achieved, are currently underway in our laboratory.
4. Materials and Methods
4.1. General Information
Proton (1H NMR), carbon (13C NMR), fluorine (19F NMR), and selenium (77Se NMR) nuclear magnetic resonance spectra were recorded on a Bruker Avance III instrument operating at 300, 400, or 500 MHz (Bruker Corporation, Billerica, MA, USA). The chemical shifts are given in parts per million (ppm) on the delta scale. The solvent peak was used as reference values for 1H NMR (CDCl3 = 7.26 ppm) and for 13C NMR (CDCl3 = 77.16 ppm). An external standard was used for 19F NMR (trifluoromethylbenzene) and for 77Se NMR (diphenyl diselenide). Data are presented as follows: Chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, m = multiplet, b = broad), integration, and coupling constants (J/Hz). High-resolution mass spectra (HRMS) data were recorded on a micrOTOF spectrometer (Bruker Corporation, Billerica, MA, USA) equipped with an orthogonal electrospray interface (ESI). Analytical thin layer chromatography (TLC plates from Merck KGaA, Darmstadt, Germany) was carried out on silica gel 60 F254 plates with visualization by ultraviolet light. Reagents and solvents were purified using standard means. Tetrahydrofuran (THF) was distilled from sodium metal/benzophenone and stored under an argon atmosphere. Anhydrous reactions were carried out in flame-dried glassware and under an argon atmosphere. Bipyridine 3 [38] and bis(perfluorophenyl)diselenide 5a [53] were prepared according to the literature. All other chemicals were used as received. An Agilent Technologies (Waldbronn, Germany) 1100 Series HPLC system (high-pressure binary gradient system equipped with a diode-array detector operating at multiple wavelengths (220, 254, 280 nm), a programmable autosampler with a 20 μL loop, and a thermostated column compartment) was employed for multimilligram separations. Data acquisition and analyses were carried out with Agilent Technologies ChemStation Version B.04.03 chromatographic data software. The UV absorbance is reported as milliabsorbance units (mAU). Chiralpak IA (immobilized amylose tris-3,5-dimethylphenylcarbamate) and Chiralpak IC (immobilized cellulose tris-3,5-dichlorophenylcarbamate) were used as chiral columns (250 × 4.6 mm) (5 μm) (Chiral Technologies Europe, Illkirch, France). HPLC-grade n-hexane (Hex) and 2-propanol (IPA) were purchased and used as received. CD spectra were recorded on a « J-810 » from Jasco (JASCO International Co. Ltd., Tokyo, Japan) at room temperature using 0.15–0.30 mM samples in ethanol and a 1 mm quartz cell, with the following conditions: 100 nm/min scanning speed, 1 nm data pitch, 4.0 nm bandwidth, 1 s response time.
4.2. General Procedure for the Synthesis of 2, 6, and 7 from Bipyridine 3
Bipyridine 3 (0.19–0.47 mmol) was dissolved in THF (5–8 mL) and the solution was cooled to −78 °C. n-BuLi (1.2 M in hexanes, 1.01 eq.) was added dropwise and the mixture was stirred at −78 °C for 1 h. A solution of diselenide 5a–c (2.5 eq.) in THF (2–5 mL) was slowly added and stirring was maintained at −78 °C for 30 min. The temperature was raised to room temperature during 5 h before addition of water (10 mL). The mixture was extracted with ethyl acetate (4 × 10 mL); the organic phases were combined, washed with brine (30 mL), and dried over anhydrous Na2SO4. After concentration, the crude was purified by chromatography on silica gel (20% dichloromethane/pentane) and, if necessary, a flash column chromatography was used to remove impurities (2% ethylacetate/cyclohexane).
5,5′-Dibromo-2,2′-dichloro-3-((perfluorophenyl)selanyl)-4,4′-bipyridine (2). Colorless oil (46 mg, 34%). Rf = 0.48 (40% dichloromethane/pentane). 1H NMR (500 MHz, CD2Cl2) δ 8.66 (s, 1H); 8.62 (s, 1H); 7.07 (s, 1H). 13C NMR (126 MHz, CD2Cl2) δ 155.1; 152.7; 152.6; 152.6; 151.2; 149.7; 148.6–145.8 (m); 144.1–141.2 (m); 139.5–136.8 (m); 126.1, 125.1 (d, J = 1.3 Hz); 120.3; 119.9; 102.6 (t, J = 23.3 Hz). 19F NMR (471 MHz, CD2Cl2) δ −127.0 to −127.1 (m); −150.7 to −150.8 (m); −159.8 to −160.0 (m). 77Se NMR (114 MHz, CD2Cl2) δ 285.2. HRMS (ESI–TOF) [M + H]+ m/z: Calcd. for C16H4Br2Cl2F5N2Se 626.7198, found: 626.7180.
5,5′-Dibromo-2,2′-dichloro-3-(phenylselanyl)-4,4′-bipyridine (6). Colorless oil (54 mg, 20%). Rf = 0.29 (5% ethylacetate/cyclohexane). 1H NMR (500 MHz, CDCl3) δ 8.62 (s, 1H); 8.54 (s, 1H); 7.31–7.26 (m, 1H); 7.21 (t, J = 7.6 Hz, 2H); 7.17–7.12 (m, 2H); 6.71 (s, 1H). 13C NMR (126 MHz, CDCl3) δ 155.5; 152.7; 152.0; 151.4; 150.3; 149.5; 132.8; 129.9; 129.7; 129.0; 128.6; 125.1; 119.7; 119.2.77Se NMR (114 MHz, CDCl3) δ 425.2. HRMS (ESI–TOF) [M + H]+ m/z: Calcd. for C16H8Br2Cl2N2Se 536.7669, found: 536.7645.
5,5′-Dibromo-2,2′-dichloro-3-(methylselanyl)-4,4′-bipyridine (7). Colorless oil (48 mg, 51%). Rf = 0.56 (5% ethylacetate/cyclohexane). 1H NMR (500 MHz, CD2Cl2) δ 8.65 (s, 1H), 8.59 (s, 1H); 7.13 (s, 1H); 2.31 (s, 3H). 13C NMR (126 MHz, CD2Cl2) δ 155.9; 152.8; 152.0; 150.9; 150.9; 150.5; 128.2; 124.6; 119.2; 118.6; 10.0. 77Se NMR (114 MHz, CDCl3) δ 459.0. HRMS (ESI-TOF) [M + H]+ m/z: Calcd. for C11H7Br2Cl2N2Se 474.7513, found: 474.7487.
4.3. General Procedure for the Copper-Mediated Synthesis of 1, 2, and 8
Under argon atmosphere, a 25 mL Schlenck tube was filled with copper powder (2 eq.) and anhydrous NMP (1–3 mL). Bis(pentafluorophenyl)dichalcogenide (1.3 eq.) was added and the mixture stirred for 3 h at room temperature. Bipyridine 3 (1 eq.) was added and the mixture was stirred overnight at a given temperature (100 or 130 °C). The reaction was allowed to cool to room temperature, diluted with diethyl ether (30 mL), and filtered. The filtrate was washed with brine (4 × 15 mL). The organic phase was dried over Na2SO4, evaporated and the residue was submitted to two successive columns chromatography on silica gel (5% diethyl ether/pentane for the first one and 2% diethyl ether/pentane for the second).
Bipyridine 1 [37] was obtained as a colorless oil (85 mg; 74%) by using the following conditions (Table 1, entry 2): Cu powder (24.8 mg, 0.39 mmol), bis(pentafluorophenyl)disuldife 4 (103.5 mg, 0.26 mmol), bipyridine 3 (100.3 mg, 0.197 mmol), at 100 °C.
Bipyridine 2 was obtained as a colorless oil (245 mg; 81%) by using the following conditions (Table 1, entry 11): Cu powder (63.5 mg, 1.0 mmol), bis(pentafluorophenyl)diselenide 5a (251 mg, 0.51 mmol), bipyridine 3 (200.5 mg, 0.394 mmol), at 100 °C.
5′-Bromo-2,2′-dichloro-3,5-bis((perfluorophenyl)thio)-4,4′-bipyridine (8) (Table 1, entry 3). It was obtained as a colorless oil (16 mg, 39%) by using the following conditions: Cu powder (9.5 mg, 0.15 mmol), bis(pentafluorophenyl) disuldife 4 (30.6 mg, 0.077 mmol), bipyridine 3 (30 mg, 0.059 mmol), at 130 °C. 1H NMR (500 MHz, CDCl3) δ 8.59 (s, 1H); 8.47 (s, 1H); 7.12 (s, 1H). 13C NMR (125 MHz, CDCl3) δ 155.2; 152.4; 152.2; 151.8; 151.0; 148.4–147.5 (m); 146.6; 146.4–144.9 (m); 140.08–135.52 (m); 128.6; 127.0, 124.6; 119.8; 106.9–105.4 (m); 106.8–105.6 (m). 19F NMR (471 MHz, CDCl3) δ −131.1 to −131.3 (m); −132.3 to −132.4 (m); −148.1 (t, J = 20.9 Hz); −150.0 (t, J = 20.8 Hz); −158.4 to −158.7 (m); −159.0 to −159.4 (m). HRMS (EI+) [M]+ m/z: Calcd. for C22H3BrCl2N2S2 698.8257, found: 698.8211.
4.4. General Procedure for the Catalytic Reduction of 2-Phenylquinoline 10
2-Phenylquinoline 10 (9.5 mg, 49 μmol), Hantzsch ester (27.1 mg, 107 μmol) and hexamethylbenzene as internal standard (δ 2.23 ppm, s, 18H), were weighted in to a screw cap vial and suspended in dry degassed CD2Cl2 (1 mL). Then, the desired amount of catalyst (5 or 20 mol%) was added from a stock solution of catalyst (120 mM). The vial was tightly sealed and stirred at 25 °C. 1H NMR spectra of aliquots of the reaction mixture (5–10 μL) diluted in CDCl3 (0.5 mL) were recorded at varying time intervals. When the reaction was finished or at 72 h maximum, the solvent was removed under reduced pressure and the remaining product was purified by flash column chromatography on silica gel (3% ethyl acetate/pentane) to afford tetrahydroquinoline 11.
4.5. Crystal Data for 2
C16H3Br2Cl2F5N2Se, M = 627.88, monoclinic, a = 10.9949(5) Å, b = 13.7036(6) Å, c = 12.1716(6) Å, V = 1827.20(15) Å3, T = 120(2) K, space group P21/c, Z = 4, μ (Mo Kα) = 6.775 mm−1, 74843 reflections measured, 6383 independent reflections (Rint = 0.0467). The final R1 value were 0.0221 (I > 2σ(I)) and 0.0240 (all data). The final wR(F2) values were 0.0552 (I > 2σ(I)) and 0.0560 (all data). The goodness of fit on F2 was 1.117. CCDC no. 1963859.
Supplementary Materials
The following are available online at https://www.mdpi.com/1420-3049/24/24/4484/s1: Figures S1 and S2: ECD spectra of 6 and 7; Figures S3 and S4: Calculated conformations for 6 and 7; Figures S5–S8: Comparison of measured and calculated UV spectra; Figures S9–S12: Calculated ECD spectra for each conformer of (M)-1, (M)-2, (M)-6 and (M)-7; Figures S13 and S14: Comparison of measured and calculated ECD spectra for (M)-6 and (M)-7; Figure S15: ORTEP plot of (P)-6; Tables S1–S5: X-Ray details for bipyridine 2; Figures S16–S18: Intermolecular interactions in the X-Ray structure of 2, DFT details; Table S6: Anion binding experiments: Overview of host addition; Table S7: Binding constants Ka for 1 and 2; Figures S19–S34: NMR spectra.
Author Contributions
Conceptualization, V.M. and P.P. (Paola Peluso); methodology, V.M. and R.W.; chemical experiments, R.W.; HPLC, P.P. (Paola Peluso); D.F.T. calculations, E.A. and P.P. (Paola Peluso); X-ray analysis, E.A.; inspiration and discussions, all authors; data curation, V.M.; writing—original draft preparation, V.M.; writing—review and editing, R.W., E.A., P.P. (Paola Peluso), S.C., P.P. (Patrick Pale) and V.M.; funding acquisition, V.M. and S.C.
Funding
This research was funded by the International Center Frontier Research in Chemistry (icFRC), the LabEx CSC (ANR-10-LABX-0026 CSC), and Ca’ Foscari University of Venice, Italy (Department of Molecular Science and Nanosystems, DSMN ADIR funds).
Acknowledgments
The authors thank the University of Strasbourg and the C.N.R.S. for their support. The CINES/CEA CCRT/IDRIS is thanked for allocation of computing time (project A0030807449). High performance computing resources were partially provided by the EXPLOR center hosted by the University of Lorraine.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Aakeroy, C.B.; Bryce, D.L.; Desiraju, G.R.; Frontera, A.; Legon, A.C.; Nicotra, F.; Rissanen, K.; Scheiner, S.; Terraneo, G.; Metrangolo, P.; et al. Definition of the chalcogen bond (IUPAC Recommendations 2019). Pure Appl. Chem. 2019, 91, 1889–1892. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Kopylovich, M.N.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Chalcogen bonding in synthesis, catalysis and design of materials. Dalton Trans. 2017, 46, 10121–10138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, L.; Wonner, P.; Huber, S.M. Chalcogen bonding: An overview. Angew. Chem. Int. Ed. 2019, 58, 1880–1891. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Lane, P.; Clark, T.; Politzer, P. Sigma-hole bonding: Molecules containing group VI atoms. J. Mol. Model. 2007, 13, 1033–1038. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [Green Version]
- Tepper, R.; Schubert, U.S. Halogen bonding in solution: Anion recognition, templated self-assembly, and organocatalysis. Angew. Chem. Int. Ed. 2018, 57, 6004–6016. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M. Halogen bonding: A halogen-centered noncovalent interaction yet to be understood. Inorganics 2019, 7, 40. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Ji, B.; Zhang, Y. Chalcogen bond: A sister noncovalent bond to halogen bond. J. Phys. Chem. A 2009, 113, 8132–8135. [Google Scholar] [CrossRef]
- Kolar, M.H.; Hobza, P. Computer modeling of halogen bonds and other σ-hole interactions. Chem. Rev. 2016, 116, 5155–5187. [Google Scholar] [CrossRef] [Green Version]
- Beno, B.R.; Yeung, K.-S.; Bartberger, M.D.; Pennington, L.D.; Meanwell, N.A. A survey of the role of noncovalent sulfur interactions in drug design. J. Med. Chem. 2015, 58, 4383–4438. [Google Scholar] [CrossRef]
- Scilabra, P.; Terraneo, G.; Resnati, G. The chalcogen bond in crystalline solids: A world parallel to halogen bond. Acc. Chem. Res. 2019, 52, 1313–1324. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.C.; Beer, P.D. Sigma-hole interactions in anion recognition. Chem 2018, 4, 731–783. [Google Scholar] [CrossRef]
- Sutar, R.; Huber, S.M. Catalysis of organic reactions through halogen bonding. ACS Catal. 2019, 9, 9622–9639. [Google Scholar] [CrossRef]
- Bamberger, J.; Ostler, F.; García Mancheño, O. Frontiers in halogen and chalcogen-bond donor organocatalysis. ChemCatChem 2019, 11. [Google Scholar] [CrossRef]
- Benz, S.; Lopez-Andarias, J.; Mareda, J.; Sakai, N.; Matile, S. Catalysis with chalcogen bonds. Angew. Chem. Int. Ed. 2017, 56, 812–815. [Google Scholar] [CrossRef]
- Benz, S.; Mareda, J.; Besnard, C.; Sakai, N.; Matile, S. Catalysis with chalcogen bonds: Neutral benzodiselenazole scaffolds with high-precision selenium donors of variable strength. Chem. Sci. 2017, 8, 8164–8169. [Google Scholar] [CrossRef] [Green Version]
- Benz, S.; Poblador-Bahamonde, A.I.; Low-Ders, N.; Matile, S. Catalysis with pnictogen, chalcogen, and halogen bonds. Angew. Chem. Int. Ed. 2018, 57, 5408–5412. [Google Scholar] [CrossRef]
- Benz, S.; Besnard, C.; Matile, S. Chalcogen-bonding catalysis: From neutral to cationic benzodiselenazole scaffolds. Helv. Chim. Acta 2018, 101, e1800075. [Google Scholar] [CrossRef]
- Wonner, P.; Vogel, L.; Kniep, F.; Huber, S.M. Catalytic carbon–chlorine bond activation by selenium-based chalcogen bond donors. Chem. Eur. J. 2017, 23, 16972–16975. [Google Scholar] [CrossRef]
- Wonner, P.; Vogel, L.; Düser, M.; Gomes, L.; Kniep, F.; Mallick, B.; Werz, D.B.; Huber, S.M. Carbon–halogen bond activation by selenium-based chalcogen bonding. Angew. Chem. Int. Ed. 2017, 56, 12009–12012. [Google Scholar] [CrossRef]
- Wonner, P.; Dreger, A.; Engelage, E.; Huber, S.M. Chalcogen bonding catalysis in a nitro-michael reaction. Angew. Chem. Int. Ed. 2019, 58, 16923–16927. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhu, H.; Liu, S.; Zhao, Z.; Zhang, L.; Hao, J.; Wang, Y. Chalcogen-chalcogen bonding catalysis enables assembly of discrete molecules. J. Am. Chem. Soc. 2019, 141, 9175–9179. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.Y.C.; Liew, J.Y.; Beer, P.D. Thermodynamics of anion binding by chalcogen bonding receptors. Chem. Eur. J. 2018, 24, 14560–14566. [Google Scholar] [CrossRef]
- Lim, J.Y.C.; Marques, I.; Félix, V.; Beer, P.D. Chiral halogen and chalcogen bonding receptors for discrimination of stereo- and geometric dicarboxylate isomers in aqueous media. Chem. Commun. 2018, 54, 10851–10854. [Google Scholar] [CrossRef]
- Benz, S.; Macchione, M.; Verolet, Q.; Mareda, J.; Sakai, N.; Matile, S. Anion transport with chalcogen bonds. J. Am. Chem. Soc. 2016, 138, 9093–9096. [Google Scholar] [CrossRef] [Green Version]
- Macchione, M.; Tsemperouli, M.; Goujon, A.; Mallia, A.R.; Sakai, N.; Sugihara, K.; Matile, S. Mechanosensitive oligodithienothiophenes: transmembrane anion transport along chalcogen-bonding cascades. Helv. Chim. Acta 2018, 101, e1800014. [Google Scholar] [CrossRef] [Green Version]
- Lee, L.M.; Tsemperouli, M.; Poblador-Bahamonde, A.I.; Benz, S.; Sakai, N.; Sugihara, K.; Matile, S. Anion transport with pnictogen bonds in direct comparison with chalcogen and halogen bonds. J. Am. Chem. Soc. 2019, 141, 810–814. [Google Scholar] [CrossRef]
- Knowles, R.R.; Jacobsen, E.N. Attractive noncovalent interactions in asymmetric catalysis: Links between enzymes and small molecule catalysts. Proc. Natl. Acad. Sci. USA 2010, 107, 20678–20685. [Google Scholar] [CrossRef] [Green Version]
- By Liu, M.; Zhang, L.; Wang, T. Supramolecular chirality in self-assembled systems. Chem. Rev. 2015, 115, 7304–7397. [Google Scholar] [CrossRef]
- Peluso, P.; Mamane, V.; Cossu, S. Liquid chromatography enantioseparations of halogenated compounds on polysaccharide-based chiral stationary phases: Role of halogen substituents in molecular recognition. Chirality 2015, 27, 667–684. [Google Scholar] [CrossRef]
- Peluso, P.; Mamane, V.; Aubert, E.; Cossu, S. Insights into the impact of shape and electronic properties on the enantioseparation of polyhalogenated 4,4’-bipyridines on polysaccharide-type selectors. Evidence for stereoselective halogen bonding interactions. J. Chromatogr. A 2014, 1345, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Peluso, P.; Mamane, V.; Aubert, E.; Dessi, A.; Dallocchio, R.; Dore, A.; Pale, P.; Cossu, S. Insights into halogen bond driven enantioseparations. J. Chromatogr. A 2016, 1467, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Peluso, P.; Mamane, V.; Aubert, E.; Cossu, S. Recent trends and applications in liquid-phase chromatography enantioseparation of atropisomers. Electrophoresis 2017, 38, 1830–1850. [Google Scholar] [CrossRef] [PubMed]
- Peluso, P.; Mamane, V.; Dallocchio, R.; Dessi, A.; Villano, R.; Sanna, D.; Aubert, E.; Pale, P.; Cossu, S. Polysaccharide-based chiral stationary phases as halogen bond acceptors: A novel strategy for detection of stereoselective σ-hole bonds in solution. J. Sep. Sci. 2018, 41, 1247–1256. [Google Scholar] [CrossRef]
- Dallocchio, R.; Dessi, A.; Solinas, M.; Arras, A.; Cossu, S.; Aubert, E.; Mamane, V.; Peluso, P. Halogen bond in high-performance liquid chromatography enantioseparations: Description, features and modelling. J. Chromatogr. A 2018, 1563, 71–81. [Google Scholar] [CrossRef]
- Peluso, P.; Dessi, A.; Dallocchio, R.; Mamane, V.; Cossu, S. Recent studies of docking and molecular dynamics simulation for liquid-phase enantioseparations. Electrophoresis 2019, 40, 1881–1896. [Google Scholar] [CrossRef]
- Peluso, P.; Gatti, C.; Dessi, A.; Dallocchio, R.; Weiss, R.; Aubert, E.; Pale, P.; Cossu, S.; Mamane, V. Enantioseparation of fluorinated 3-arylthio-4,4’-bipyridines: insights into chalcogen and π-hole bonds in high-performance liquid chromatography. J. Chromatogr. A 2018, 1567, 119–129. [Google Scholar] [CrossRef]
- Mamane, V.; Aubert, E.; Peluso, P.; Cossu, S. Lithiation of prochiral 2,2’-dichloro-5,5’-dibromo-4,4’-bipyridine as a tool for the synthesis of chiral polyhalogenated 4,4’-bipyridines. J. Org. Chem. 2013, 78, 7683–7689. [Google Scholar] [CrossRef]
- Mamane, V.; Aubert, E.; Peluso, P.; Cossu, S. Synthesis, resolution, and absolute configuration of chiral 4,4′-bipyridines. J. Org. Chem. 2012, 77, 2579–2583. [Google Scholar] [CrossRef]
- Kondratenko, N.V.; Kolomeytsev, A.A.; Popov, V.I.; Yagupolskii, L.M. Synthesis and reactions of trifluoromethylthio(seleno)- and pentafluorophenylthio(seleno)-copper. Synthesis 1985, 667–669. [Google Scholar] [CrossRef]
- Haupt, A.; Lentz, D. Tuning the electron affinity and stacking properties of corannulene by introduction of fluorinated thioethers. Chem. Asian J. 2018, 13, 3022–3026. [Google Scholar] [CrossRef] [PubMed]
- Mamane, V.; Peluso, P.; Aubert, E.; Cossu, S.; Pale, P. Chiral hexahalogenated 4,4′-bipyridines. J. Org. Chem. 2016, 81, 4576–4587. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.Q.; Zhao, X.R.; Wang, H.; Jin, W.J. The Competition of σ-Hole·Cl− and π-Hole·Cl− bonds between C6F5X (X = F, Cl, Br, I) and the chloride anion and its potential application in separation science. J. Phys. Chem. B 2014, 118, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Thordarson, P. Determining association constants from titration experiments in supramolecular chemistry. Chem. Soc. Rev. 2011, 40, 1305–1323. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; You, S.-L. Transfer hydrogenation with Hantzsch esters and related organic hydride donors. Chem. Soc. Rev. 2012, 41, 2498–2518. [Google Scholar] [CrossRef] [PubMed]
- Rueping, M.; Antonchick, A.P.; Theissmann, T. A Highly enantioselective Brønsted acid catalyzed cascade reaction: Organocatalytic transfer hydrogenation of quinolines and their application in the synthesis of alkaloids. Angew. Chem. Int. Ed. 2006, 45, 3683–3686. [Google Scholar] [CrossRef]
- Bruckmann, A.; Pena, M.A.; Bolm, C. Organocatalysis through halogen-bond activation. Synlett 2008, 6, 900–902. [Google Scholar] [CrossRef]
- He, W.; Ge, Y.-C.; Tan, C.-H. Halogen-bonding-induced hydrogen transfer to C = N bond with Hantzsch ester. Org. Lett. 2014, 16, 3244–3247. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Uno, H.; Tokunaga, E.; Shibata, N. Fluorobissulfonylmethyl iodides: An efficient scaffold for halogen bonding catalysts with an sp3-hybridized carbon–iodine moiety. ACS Catal. 2018, 8, 6601–6605. [Google Scholar] [CrossRef]
- Nayak, S.K.; Kumar, V.; Murray, J.S.; Politzer, P.; Terraneo, G.; Pilati, T.; Metrangolo, P.; Resnati, G. Fluorination promotes chalcogen bonding in crystalline solids. CrystEngComm 2017, 19, 4955–4959. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. The bright future of unconventional σ/π-hole interactions. ChemPhysChem 2015, 16, 2496–2517. [Google Scholar] [CrossRef] [PubMed]
- Ser, C.T.; Yang, H.; Wong, M.W. Iodoimidazolinium-catalyzed reduction of quinoline by Hantzsch ester: Halogen bond or Brønsted acid catalysis. J. Org. Chem. 2019, 84, 10338–10348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klapötke, T.M.; Krumm, B.; Polborn, K. Synthesis, chemistry, and characterization of perfluoroaromatic selenium derivatives. Eur. J. Inorg. Chem. 1999, 8, 1359–1366. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1, 2, 6 and 7 are available from the authors. |
Figure 1.
Halogen and chalcogen bonds (EWG: Electron withdrawing group, BL: Lewis base).

Figure 2.
Chiral 3-chalcogeno-4,4′-bipyridines considered in this work.

Scheme 1.
Synthesis of bipyridine derivatives 1, 2, 6, and 7 through a lithiated intermediate.

Figure 3.
Electronic circular dichroism (ECD) spectra of the enantiomers of bipyridines 1 and 2.

Figure 4.
The different conformations calculated for bipyridine 2.

Figure 5.
Comparison of measured and calculated ECD spectra for (M)-1 and (M)-2.

Figure 6.
ORTEP plot of 2 with ellipsoids drawn at the 50% probability level.

Figure 7.
19F NMR traces of bipyridines 1 (left) and 2 (right) upon addition of TBACl.

Figure 8.
Chemical shift change (Δδ) of para-F in bipyridines 1 and 2 upon addition of TBACl.

Figure 9.
Ab initio electrostatic potential surfaces (EPS) of bipyridines 6, 1, and 2.

Scheme 2.
Possible mechanism for the ChB-catalyzed reduction of 2-phenylquinoline 10.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Synthesis of 1 and 2 through copper-mediated cross-coupling reaction.

| Entry | ArChChAr | n (eq.) | T (°C) | NMR Ratio 3/1/8 or 3/2 | Yield (%) |
|---|---|---|---|---|---|
| 1 | (C6F5S)2 (4) | 1.3 | 80 | 33/67/0 | − |
| 2 | ‘’ | 1.3 | 100 | 12/77/11 | 74 (1) |
| 3 | ‘’ | 1.3 | 130 | 4/31/65 | 39 (8) |
| 4 | ‘’ | 0.5 | 100 | 78/20/2 | − |
| 5 | ‘’ | 0.8 | 100 | 53/43/4 | − |
| 6 | ‘’ | 1.0 | 100 | 39/57/4 | − |
| 7 | ‘’ | 2.0 | 100 | 0/75/25 | 67 (1) |
| 8 | ‘’ | 4.0 | 100 | 0/54/46 | |
| 9 | ‘’ | 0.5 | 130 | 49/42/9 | − |
| 10a | ‘’ | 1.3 | 100 | 17/63/20 | − |
| 11 | (C6F5Se)2 (5a) | 1.3 | 100 | 14/86 | 81 (2) |
a The reagents were mixed together without preforming the copper complex.
Table 2.
Optimized multimilligram enantioseparation of 4,4′-bipyridines 2, 6, and 7 a.
| Bipyridine | Racemate [mg] | Absolute Configuration (e.e.b %) | Recovered Amounts [mg] (Recovery%) | ||
|---|---|---|---|---|---|
| 1st Eluted Peak | 2nd Eluted Peak | 1st Eluted Peak | 2nd Eluted Peak | ||
| 2 | 30.0 | >99 | 98.8 | 10.6 (70.7) | 13.0 (86.7) |
| 6 | 40.0 | >99 | 95.0 | 7.4 (37.0) | 18.0 (90.0) |
| 7 | 12.0 | >99 | 95.1 | 5.1 (85.0) | 5.9 (98.3) |
a Mobile Phase: n-Hexane/2-propanol 90:10, flow rate 0.5 mL/min, T = 22 °C. b Enantiomeric excess (e.e.) determined by chiral HPLC under the same conditions used for recoveries.
Table 3.
Relative energies and populations of the different conformers of 1 and 2.
| 1 | 2 | |||||||
|---|---|---|---|---|---|---|---|---|
| A1 | A2 | B1 | B2 | A1 | A2 | B1 | B2 | |
| ΔG (kJ/mol) | 0.66 | 0 | 0.74 | 8.09 | 0 | 3.17 | 0.96 | 7.53 |
| Pop. (%) | 30 | 39 | 29 | 2 | 50 | 14 | 34 | 2 |
Table 4.
Effect of ChB donors 1 and 2 and reference compounds on the catalytic reduction of 10 by HE.
Table 4.
Effect of ChB donors 1 and 2 and reference compounds on the catalytic reduction of 10 by HE.

| Entry | Cat. | x (mol%) | Time (h) | Conv. (%)a | Yield (%) |
|---|---|---|---|---|---|
| 1 | 1 | 20 | 48 | 100 | 87 |
| 2 | 2 | 20 | 48 | 100 | 94 |
| 3 | 1 | 5 | 72 | 81 | − |
| 4 | 2 | 5 | 72 | 100 | 88 |
| 5 | 2 | 5 | 18 | 54 | − |
| 6 | - | - | 72 | 0 | − |
| 7b | 2 | 5 | 18 | 38 | − |
| 8 | 12 | 20 | 72 | 62 | − |
| 9 | 13 | 20 | 72 | 78 | − |
| 10 | 3 | 20 | 48 | 68 | − |
| 11 | 3 | 20 | 72 | 100 | 86 |
| 12 | (P)-2 | 20 | 48 | 100 | 90 (0% ee) |
a Calculated by 1H NMR with hexamethylbenzene as internal standard. b 1 eq. of TBACl was added.
Table 5.
Electrostatic potential (EP) maxima [kJ/mol] on Ch σ-holes for compounds 1, 2, and 6 a.
| σ-hole b | 6 | 1 | 2 |
| 1 | 88.0 | 113.2 | 136.7 |
| 2 | 73.6 | 98.6 | 127.5 |
a Spartan ’10 Version 1.1.0 (Wavefunction Inc., Irvine, CA), isovalue 0.002. b σ-hole (1) is located on the elongation of the C–Ch bond; σ-hole (2) is located on the elongation of the CArF–Ch bond.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Weiss, R.; Aubert, E.; Peluso, P.; Cossu, S.; Pale, P.; Mamane, V. Chiral Chalcogen Bond Donors Based on the 4,4′-Bipyridine Scaffold. Molecules 2019, 24, 4484. https://doi.org/10.3390/molecules24244484
AMA Style
Weiss R, Aubert E, Peluso P, Cossu S, Pale P, Mamane V. Chiral Chalcogen Bond Donors Based on the 4,4′-Bipyridine Scaffold. Molecules. 2019; 24(24):4484. https://doi.org/10.3390/molecules24244484
Chicago/Turabian StyleWeiss, Robin, Emmanuel Aubert, Paola Peluso, Sergio Cossu, Patrick Pale, and Victor Mamane. 2019. "Chiral Chalcogen Bond Donors Based on the 4,4′-Bipyridine Scaffold" Molecules 24, no. 24: 4484. https://doi.org/10.3390/molecules24244484