
Improved Process for the Synthesis of 3-(3-Trifluoromethylphenyl)propanal for More Sustainable Production of Cinacalcet HCl †
 , ,
, ,
Abstract
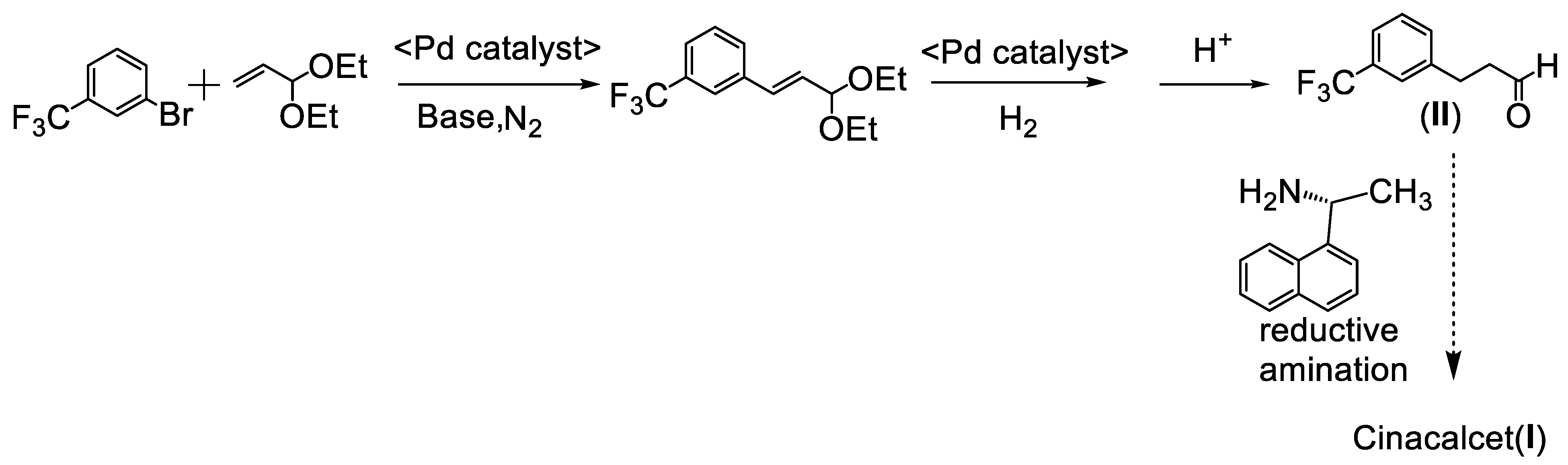
:1. Introduction
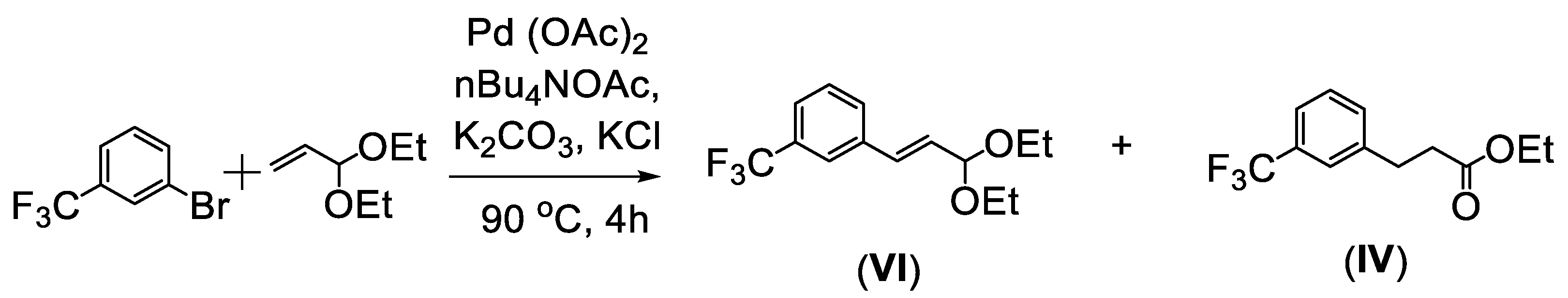
2. Mizoroki–Heck Cross-Coupling Reaction between 1-Bromo-3-(trifluoromethyl)benzene and Acroleine Diethyl Acetal
3. Hydrogenation of VI, in a Mixture with IV, by Utilizing the Same Mizoroki–Heck Catalyst Followed by Hydrolysis of 1-(3,3-Diethoxypropyl)-3-(trifluoromethyl)benzene (III) to Afford II in a Mixture with IV
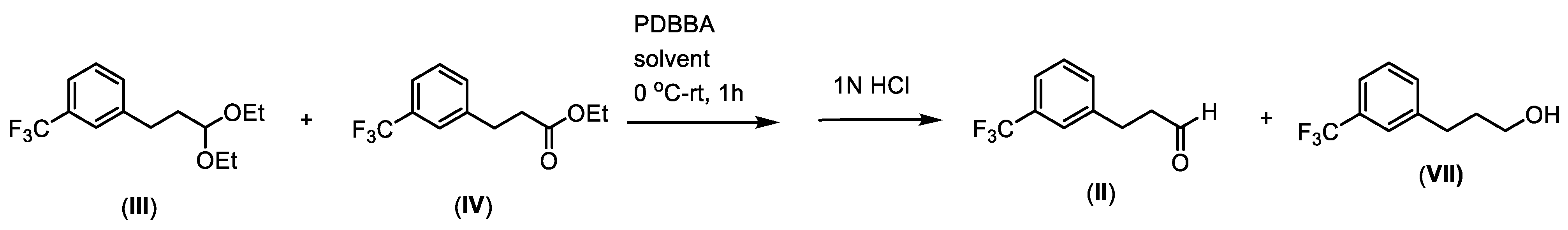
4. Purification of 3-(3-(Trifluoromethyl)phenyl)propanal (II) via Bisulfite Adduct and Regeneration of the Aldehyde
5. Reduction of Ethyl 3-(3-(Trifluoromethyl)phenyl)propanoate by-Product (IV) to 3-(3-(Trifluoromethyl)phenyl)propanal (II)
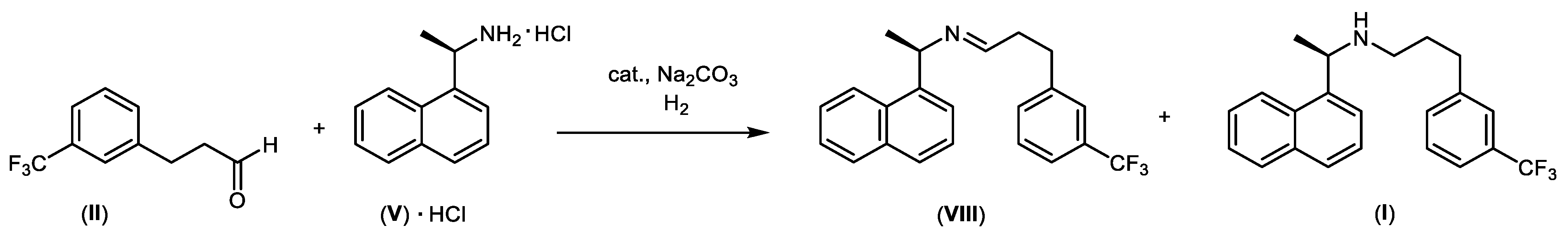
6. Synthesis of Cinacalcet Hydrochloride under Microwave Conditions
7. Experimental Section
7.1. Materials and Reagents
7.2. Characterizations and Measurements
7.3. Syntheses Using Conventional Heating
- General procedure for the synthesis of the mixture of 1-((E)-3,3-diethoxyprop-1-enyl)-3-(trifluoromethyl)benzene (VI) and ethyl 3-(3-(trifluoromethyl)phenyl)propanoate (IV)
- General procedure for the synthesis of the mixture of 1-(3,3-diethoxypropyl)-3-(trifluoromethyl)benzene (III) and ethyl 3-(3-(trifluoromethyl)phenyl)propanoate (IV)
- Acid treatment of the mixture of 1-(3,3-diethoxypropyl)-3-(trifluoromethyl)benzene (III) and ethyl 3-(3-(trifluoromethyl)phenyl)propanoate (IV)
- Purification of the mixture of 1-(3,3-diethoxypropyl)-3-(trifluoromethyl)benzene (III) and ethyl 3-(3-(trifluoromethyl)phenyl)propanoate (IV) through Bertagnini salt formation
- Procedure for the regeneration of 3-(3-(trifluoromethyl)phenyl)propanal (II) from sodium 1-hydroxy-3-(3-trifluoromethylphenyl)propane-1-sulfonate
- Procedure for the synthesis of 3-(3-(trifluoromethyl)phenyl)propanal (II) via selective reduction with potassium diisobutyl-tert-butoxyaluminum hydride (PDBBA) of the mixture of 1-(3,3-diethoxypropyl)-3-(trifluoromethyl)benzene (III) and ethyl 3-(3-(trifluoromethyl)phenyl)propanoate (IV)
- General procedure for the synthesis of 3-(3-(trifluoromethyl)phenyl)-N-((R)-1-(naphthalen-1-yl)ethyl)propan-1-amine (I) in the presence of low-metal-content catalysts
- Procedure for the preparation of the hydrochloride salt of 3-(3-(trifluoromethyl)phenyl)-N-((R)-1-(naphthalen-1-yl)ethyl)propan-1-amine (I)
8. Conclusions
- A cascade reaction was applied for the Mizoroki–Heck cross-coupling and the following hydrogenation step using the same palladium species as a catalyst, simply changing the gas atmosphere and using greener solvents than DMF;
- Palladium species were also recovered in a simple way at the end of both reactions, being described in point (a) as a heterogeneous catalyst on alumina that has been used in the reductive amination step;
- Two different protocols were applied to purify compound II using a Bertagnini salt approach or selective reduction under very mild conditions of by-product IV, which was transformed with a high yield and purity into compound II;
- Reductive amination with recyclable heterogeneous catalysts, prepared using a simple protocol developed by us, was efficiently used;
- The preparation of the API described in this paper is, in our opinion, very efficient and potentially suitable for industrial production;
- Preliminary results using MW permitted a reduction in the reaction times of some steps in the synthesis of the API, even if idoneous equipment will be required for future scaled-up investigations.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Available online: https://access.cortellis.com/login?referrer=%2Fgenerics%2F&app=generics (accessed on 8 August 2023).
- Van Wagenen, B.C.; Moe, S.T.; Balandrin, M.F.; Delmar, E.G.; Nemeth, E.F. Calcium Receptor-Active Compounds. WO9612697, 2 May 1996. [Google Scholar]
- Palmer, S.C.; Nistor, I.; Craig, J.C.; Pellegrini, F.; Messa, P.; Tonelli, M.; Covic, A.; Strippoli, F.G. Cinacalcet in Patients with Chronic Kidney Disease: A Cumulative Meta-Analysis of Randomized Controlled Trials. PLoS Med. 2013, 10, e1001436. [Google Scholar] [CrossRef] [Green Version]
- Verheyen, N.; Pilz, S.; Eller, K.; Kienreich, K.; Fahrleitner-Pammer, A.; Pieske, B.; Ritz, E.; Tomaschitz, A. Cinacalcet hydrochloride for the treatment of hyperparathyroidism. Expert Opin. Pharmacother. 2013, 14, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Barniol-Xicota, M.; Leiva, R.; Escolano, C.; Vzquez, S. Syntheses of Cinacalcet: An Enantiopure Active Pharmaceutical Ingredient (API). Synthesis 2016, 48, 783–803. [Google Scholar]
- Li, M.; Jin, Y.; Chen, Y.; Wu, W.; Jiang, H. Palladium-Catalyzed Oxidative Amination of Unactivated Olefins with Primary Aliphatic Amines. J. Am. Chem. Soc. 2023, 145, 9448–9453. [Google Scholar] [CrossRef]
- Thakore, R.R.; Takale, B.S.; Casotti, G.; Gao, E.S.; Jin, H.S.; Lipshutz, B.H. Chemoselective Reductive Aminations in Aqueous Nanoreactors Using Parts per Million Level Pd/C Catalysis. Org. Lett. 2020, 22, 6324–6329. [Google Scholar] [CrossRef] [PubMed]
- Bereczki, L.; Bombicz, P.; Bálint, C.J.; Egri, G.; Schindler, J.; Pokol, G.; Fogassy, E.; Marthi, K. Optical resolution of 1-(1-naphthyl)ethylamine by its dicarboxylic acid derivatives: Structural features of the oxalic acid derivative diastereomeric salt pair. Chirality 2009, 21, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhang, C.; He, Q.; Qin, H.; Liang, G.; Liu, W. Efficient resolution of (R,S)-1-(1-naphthyl)ethylamine by Candida antarctica lipase B in ionic liquids. Mol. Catal. 2018, 448, 116–121. [Google Scholar] [CrossRef]
- Pan, Z.; Shen, L.; Song, D.; Xie, Z.; Ling, F.; Zhong, W. B(C6F5)3-Catalyzed Asymmetric Reductive Amination of Ketones with Ammonia Borane. J. Org. Chem. 2018, 83, 11502–11509. [Google Scholar] [CrossRef]
- Marx, L.; Ríos-Lombardía, N.; Farnberger, J.F.; Kroutil, W.; Benítez-Mateos, A.I.; López-Gallego, L.; Morís, F.; González-Sabín, J.; Berglunda, P. Chemoenzymatic Approaches to the Synthesis of the Calcimimetic Agent Cinacalcet Employing Transaminases and Ketoreductases. Adv. Synth. Catal. 2018, 360, 2157–2165. [Google Scholar] [CrossRef]
- Parodi, E.; Piccolo, O.; Petri, A. Enantioselective Synthesis of Industrially Relevant Amines Using an Immobilised ω-Transaminase. In Applied Biocatalysis: The Chemist’s Enzyme Toolbox; Whittall, J., Sutton, P.W., Eds.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2020; Chapter IV; pp. 178–182. [Google Scholar]
- Berthiol, F.; Doucet, H.; Santelli, M. Direct synthesis of cinnamaldehyde derivatives by reaction of aryl bromides with 3,3-diacetoxypropene catalyzed by a palladium–tetraphosphine complex. Catal. Lett. 2005, 102, 281–284. [Google Scholar] [CrossRef]
- Battistuzzi, G.; Cacchi, S.; Fabrizi, G. An Efficient Palladium-Catalyzed Synthesis of Cinnamaldehydes from Acrolein Diethyl Acetal and Aryl Iodides and Bromides. Org. Lett. 2003, 5, 777–780. [Google Scholar] [CrossRef]
- Available online: https://nj.gov/health/eoh/rtkweb/documents/fs/0759.pdf (accessed on 8 August 2023).
- Al-Maksoud, W.; Menuel, S.; Jahjah, M.; Monflier, E.; Pinel, C.; Djakovitch, L. Base directed palladium catalysed Heck arylation of acrolein diethyl acetal in water. Appl. Catal. A Gen. 2014, 469, 250–258. [Google Scholar] [CrossRef]
- IP.com. No-IPCOM000191229D. Process for Preparing 3-(3-Trifluoromethylphenyl) Propanal. 22 December 2009. Available online: https://ip.com/ (accessed on 8 August 2023).
- Smith, M.B. March’s Advanced Organic Chemistry, 7th ed.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2013; pp. 1096–1099. [Google Scholar]
- Barniol-Xicota, M.; Turcu, A.L.; Codony, S.; Escolano, C.; Vázquez, S. Direct reductive alkylation of amine hydrochlorides with aldehyde bisulfite adducts. Tetrahedron Lett. 2014, 55, 2548–2550. [Google Scholar] [CrossRef]
- Li, X.; Iyer, K.S.; Thakore, R.R.; Leahy, D.H.; Bailey, J.D.; Lipshutz, B.H. Bisulfite Addition Compounds as Substrates for Reductive Aminations in Water. Org. Lett. 2021, 23, 7205–7208. [Google Scholar] [CrossRef]
- Kjell, D.P.; Slattery, B.J.; Semo, M.J. A Novel, Nonaqueous Method for Regeneration of Aldehydes from Bisulfite Adducts. J. Org. Chem. 1999, 64, 5722–5724. [Google Scholar] [CrossRef] [PubMed]
- Chae, M.J.; Song, J.I.; An, D.K. Chemoselective Reduction of Esters to Aldehydes by Potassium Diisobutyl-t-butoxyaluminum Hydride (PDBBA). Bull. Korean Chem. Soc. 2007, 28, 2517–2518. [Google Scholar] [CrossRef]
- Paganelli, S.; Angi, A.; Pajer, N.; Piccolo, O. A smart heterogeneous catalyst for efficient, chemo- and stereoselective hydrogenation of 3-Hexyn-1-ol. Catalysts 2021, 11, 14. [Google Scholar] [CrossRef]
- Paganelli, S.; Tassini, R.; Rathod, V.D.; Onida, O.; Fiorilli, S.; Piccolo, O. A low Rhodium content smart catalyst for hydrogenation and hydroformylation reactions. Catal. Lett. 2020, 151, 1508–1521. [Google Scholar] [CrossRef]
- Airoldi, V.; Piccolo, O.; Roda, G.; Appiani, R.; Bavo, F.; Tassini, R.; Paganelli, S.; Arnoldi, S.; Pallavicini, M.; Bolchi, C. Efficient One-Pot Reductive Aminations of Carbonyl Compounds with Aquivion-Fe as a Recyclable Catalyst and Sodium Borohydride. Eur. J. Org. Chem. 2020, 2, 162–168. [Google Scholar] [CrossRef]
- Kranjc, K.; Kočevar, M. From conventional reaction conditions to microwave-assisted catalytic transformations of various substrates. State of the art in 2012 (Part A: General). Curr. Org. Chem. 2013, 17, 448–456. [Google Scholar] [CrossRef]
- Kranjc, K.; Kočevar, M. From conventional reaction conditions to microwave-assisted catalytic transformations of various substrates. State of the art in 2012 (Part B: Catalysis). Curr. Org. Chem. 2013, 17, 457–473. [Google Scholar] [CrossRef]
- Gawande, M.B.; Shelke, S.N.; Zboril, R.; Varma, R.S. Microwave-Assisted Chemistry: Synthetic Applications for Rapid Assembly of Nanomaterials and Organics. Acc. Chem. Res. 2014, 47, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- Martina, K.; Cravotto, G.; Varma, R.S. Impact of Microwaves on Organic Synthesis and Strategies toward Flow Processes and Scaling Up. J. Org. Chem. 2021, 86, 13857–13872. [Google Scholar] [PubMed]
- Irfan, M.; Fuchs, M.; Glasnov, T.N.; Kappe, C.O. Microwave-Assisted Cross-Coupling and Hydrogenation Chemistry by Using Heterogeneous Transition-Metal Catalysts: An Evaluation of the Role of Selective Catalyst Heating. Chem. Eur. J. 2009, 15, 11608–11618. [Google Scholar] [CrossRef]
- Ganesh, N.S.; Spandana, K.V.L.D.; Narsaiah, A.V. Enantioselective synthesis of hyperparathyroidism agent Cinacalcet hydrochloride. Indian J. Chem. 2022, 61, 503–507. [Google Scholar]
- Allegrini, P.; Attolino, E.; Rossi, D. A Process for the Preparation of Cinacalcet and Intermediates Thereof. EP2327684, 7 October 2015. [Google Scholar]
- Chavakula, R.; Mutyala, N.R.; Chennupati, S.R. Industrially viable synthesis of 3-[3-(trifluoromethyl)phenyl]propionaldehyde.A key intermediate of cinacalcet. J. Indian Chem. Soc. 2013, 90, 1259–1261. [Google Scholar]
- Hewitt, K.A.; Herbert, C.A.; Jarvo, E.R. Synthesis of Vicinal Carbocycles by Intramolecular Nickel-Catalyzed Conjunctive Cross-Electrophile Coupling Reaction. Org. Lett. 2022, 24, 6093–6098. [Google Scholar] [CrossRef]
- IP.com. No-000135782D. Isolated (R) α Methyl N-[3-[3-(trifluoromethyl)phenyl]propyl]-1-naphthalenemethane Amine. 23 April 2006. Available online: https://ip.com/ (accessed on 8 August 2023).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run a | Ligand | Solvent | Base | Additive | Conv. [%] | VI Yield % | IV Yield % |
|---|---|---|---|---|---|---|---|
| 1 | nBu4NOAc | DMF | K2CO3 | KCl | 100 | 85 | 15 |
| 2 | nBu4NOAc | DMF | K2CO3 | - | 100 | 80 | 20 |
| 3 | nBu4NOAc | γ-valerolactone | K2CO3 | KCl | 100 | 95 | 5 |
| 4 | nBu4NOAc | 2-Me-THF | K2CO3 | KCl | 100 | 93 | 7 |
| 5 | nBu4NOAc | CPME | K2CO3 | KCl | 100 | 80 | 20 |
| 6 | nBu4NBr | DMF | K2CO3 | KCl | 100 | 40 | 60 |
| 7 | nBu4NBr | DMF | LiOH.H2O | KCl | 100 | 60 | 40 |
| 8 | nBu4NBr | DMF | DBU | - | 58 | 58 | - |
| 9 | nBu4NBr | DMF | NaOMe | - | 1 | - | 1 |
| 10 | nBu4NBr | DMF | Cs2CO3 | - | 29 | 7 | 22 |
| 11 | nBu4NBr | DMF | Et3N | - | 66 | 1 | 65 |
| 12 | nBu4NBr | DMF | DCHMA | - | 95 | 50 | 45 |
| 13 b | nBu4NOAc | DMF | K2CO3 | KCl | 100 | 99 | 1 |
| Run a | Reagent | Molar amount Based to IV | T (°C)/ t (h) | Solvent | Conv. of IV [%] | II [%] | VII [%] |
|---|---|---|---|---|---|---|---|
| 1 | PDBBA | 1.3 | 0–25/4 | Toluene | 100 | 96 | 4 |
| 2 | PDBBA | 1.3 | 0–25/4 | DCM | 100 | 92 | 8 |
| 3 | PDBBA | 1.3 | 0–25/4 | THF | 100 | 90 | 10 |
| 4 b | PDBBA | 1.25 | 0–25/1 | Toluene | 100 | 97 | 3 |
| 5 | SDBBA | 1.3 | 0–25/4 | Toluene | 100 | 90 | 10 |
| 6 | SDBBA | 1.3 | 0–25/4 | DCM | 100 | 85 | 15 |
| 7 c | DIBALH | 1.1 | −75/1 | THF | 100 | 60 | 40 |
| Run | Catalyst | Conv. [%] | VIII % | I % |
|---|---|---|---|---|
| 1 a | 0.28% Pd/Al2O3 | 100 | 2 | 98 |
| 2 b | 0.28% Pd/Al2O3 | 100 | 1 | 99 |
| 3 b | 0.28% Pd/Al2O3 | 100 | 1 | 99 |
| 4 b | 0.28% Pd/Al2O3 | 100 | 3 | 97 |
| 5 c | 0.18% Rh/Al2O3 | 100 | 38 | 68 |
| 6 d | 0.18% Pd/Al2O3 | 100 | 3 | 97 |
| 7 b,d | 0.18% Pd/Al2O3 | 100 | 3 | 97 |
| 8 b,d | 0.18% Pd/Al2O3 | 100 | 3 | 97 |
| 9 b,d | 0.18% Pd/Al2O3 | 100 | 4 | 96 |
| 10 b,d | 0.18% Pd/Al2O3 | 100 | 4 | 96 |
| Run a | Solvent | Conv. (%) | VI (%) | IV (%) |
|---|---|---|---|---|
| 1 | 2-Me-THF | 100 | 92 | 8 |
| 2 | γ-Valerolactone | 100 | 91 | 9 |
| 3 | THF | 100 | 90 | 10 |
| Run a | Conv. (%) | Time (h) | III (%) | IV (%) |
|---|---|---|---|---|
| 1 | 91 | 2 | 91 | 8 |
| 2 | 100 | 4 | 92 | 8 |
| Run a | T (°C)/t (h) | Conv.% | VIII% | I% |
|---|---|---|---|---|
| 1 | 60/8 | 100 | - | 100 |
| 2 b | 60/8 | 100 | 1 | 99 |
| 3 c | 60/8 | 100 | 48 | 42 |
| 4 d | 60/8 | 100 | 2 | 98 |
| 5 e | 60/8 | 100 | 52 | 48 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rathod, V.D.; Paganelli, S.; Kočevar, M.; Krivec, M.; Piccolo, O. Improved Process for the Synthesis of 3-(3-Trifluoromethylphenyl)propanal for More Sustainable Production of Cinacalcet HCl. Molecules 2023, 28, 6042. https://doi.org/10.3390/molecules28166042
Rathod VD, Paganelli S, Kočevar M, Krivec M, Piccolo O. Improved Process for the Synthesis of 3-(3-Trifluoromethylphenyl)propanal for More Sustainable Production of Cinacalcet HCl. Molecules. 2023; 28(16):6042. https://doi.org/10.3390/molecules28166042
Chicago/Turabian StyleRathod, Vikas Damu, Stefano Paganelli, Marijan Kočevar, Marko Krivec, and Oreste Piccolo. 2023. "Improved Process for the Synthesis of 3-(3-Trifluoromethylphenyl)propanal for More Sustainable Production of Cinacalcet HCl" Molecules 28, no. 16: 6042. https://doi.org/10.3390/molecules28166042