
Platinum(0)-η2-1,2-(E)ditosylethene Complexes Bearing Phosphine, Isocyanide and N-Heterocyclic Carbene Ligands: Synthesis and Cytotoxicity towards Ovarian and Breast Cancer Cells

 , ,
, ,
Abstract
1. Introduction
2. Results
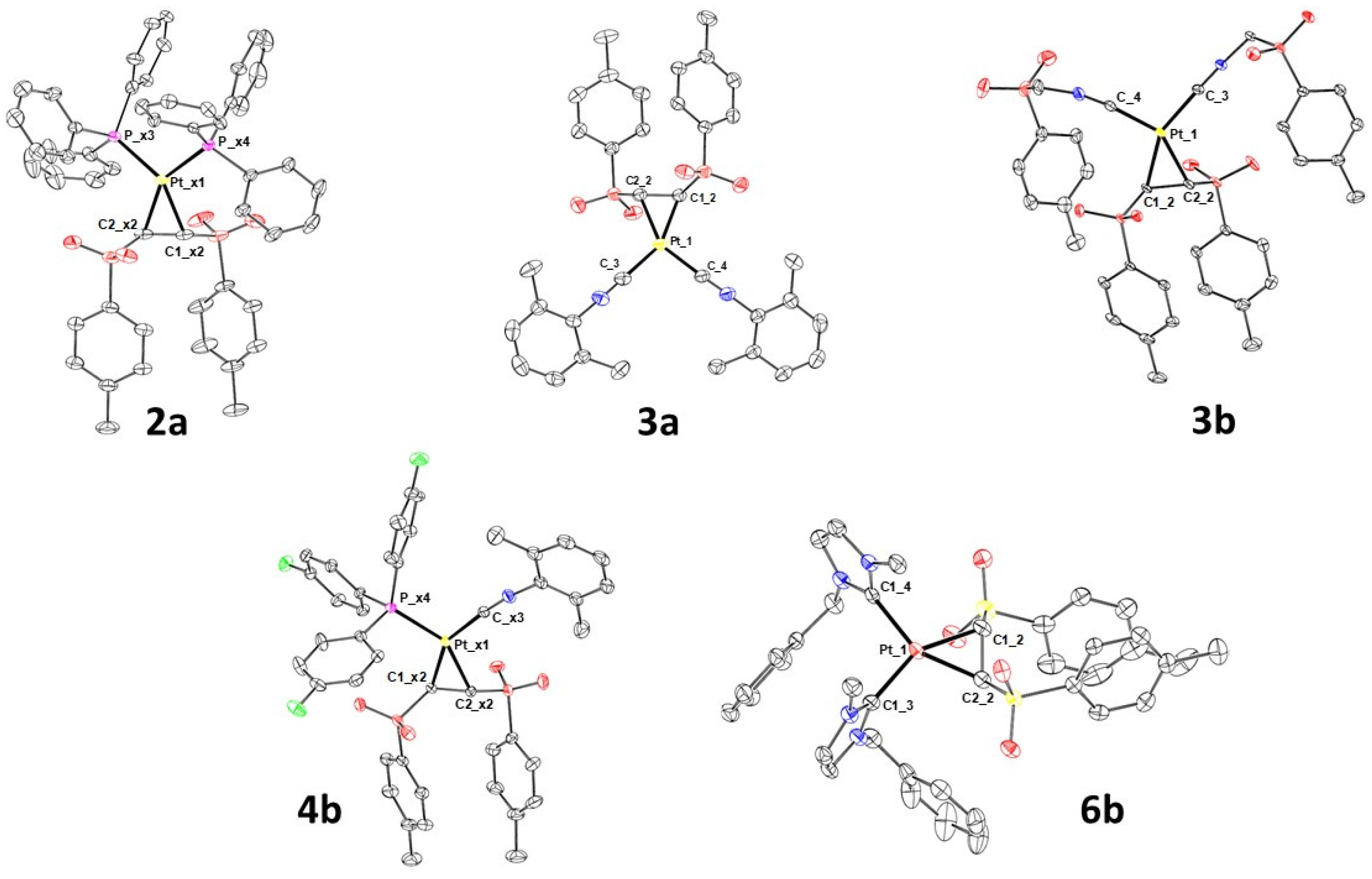
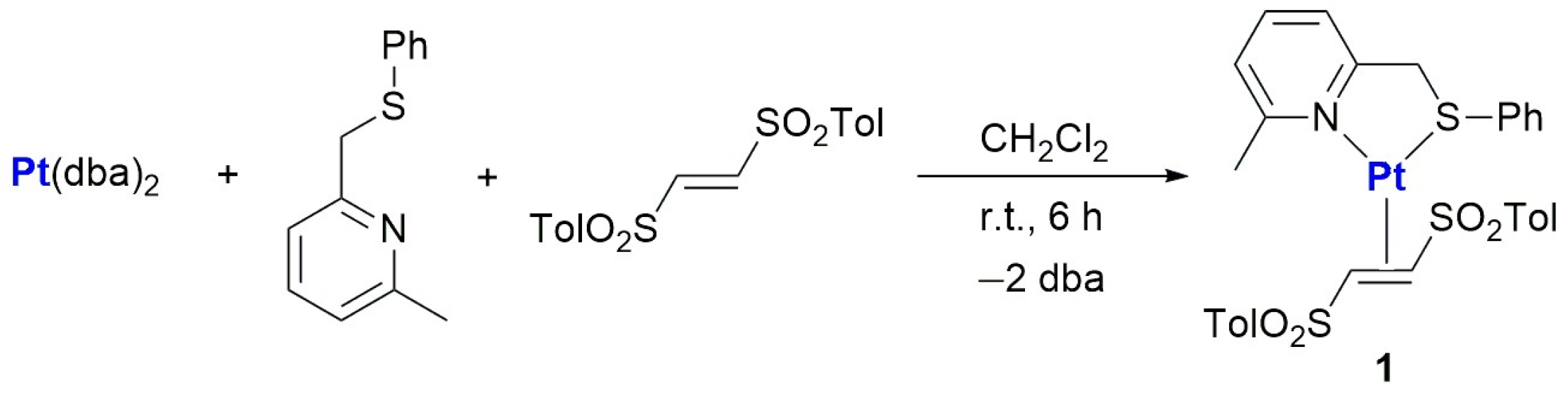
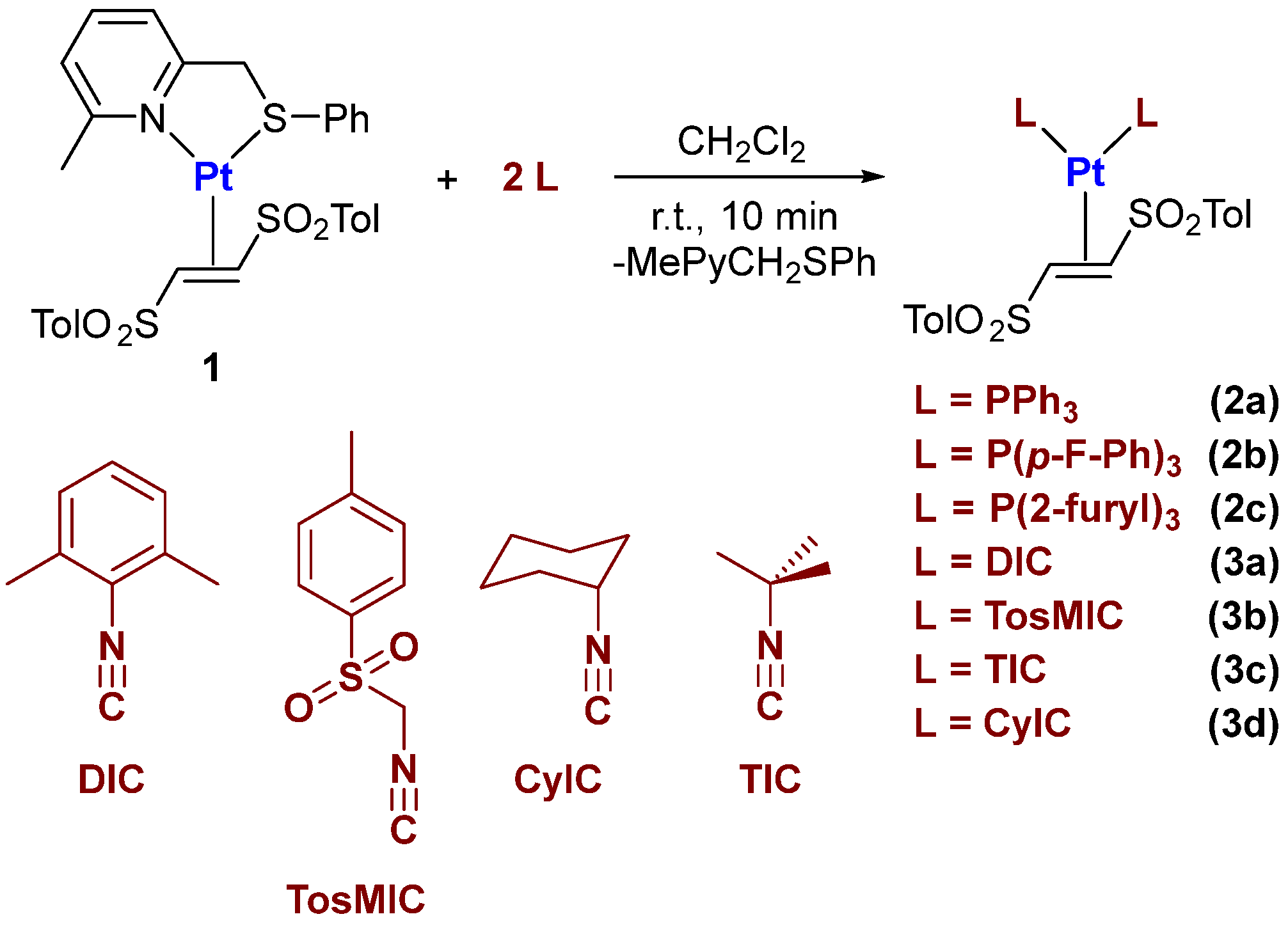
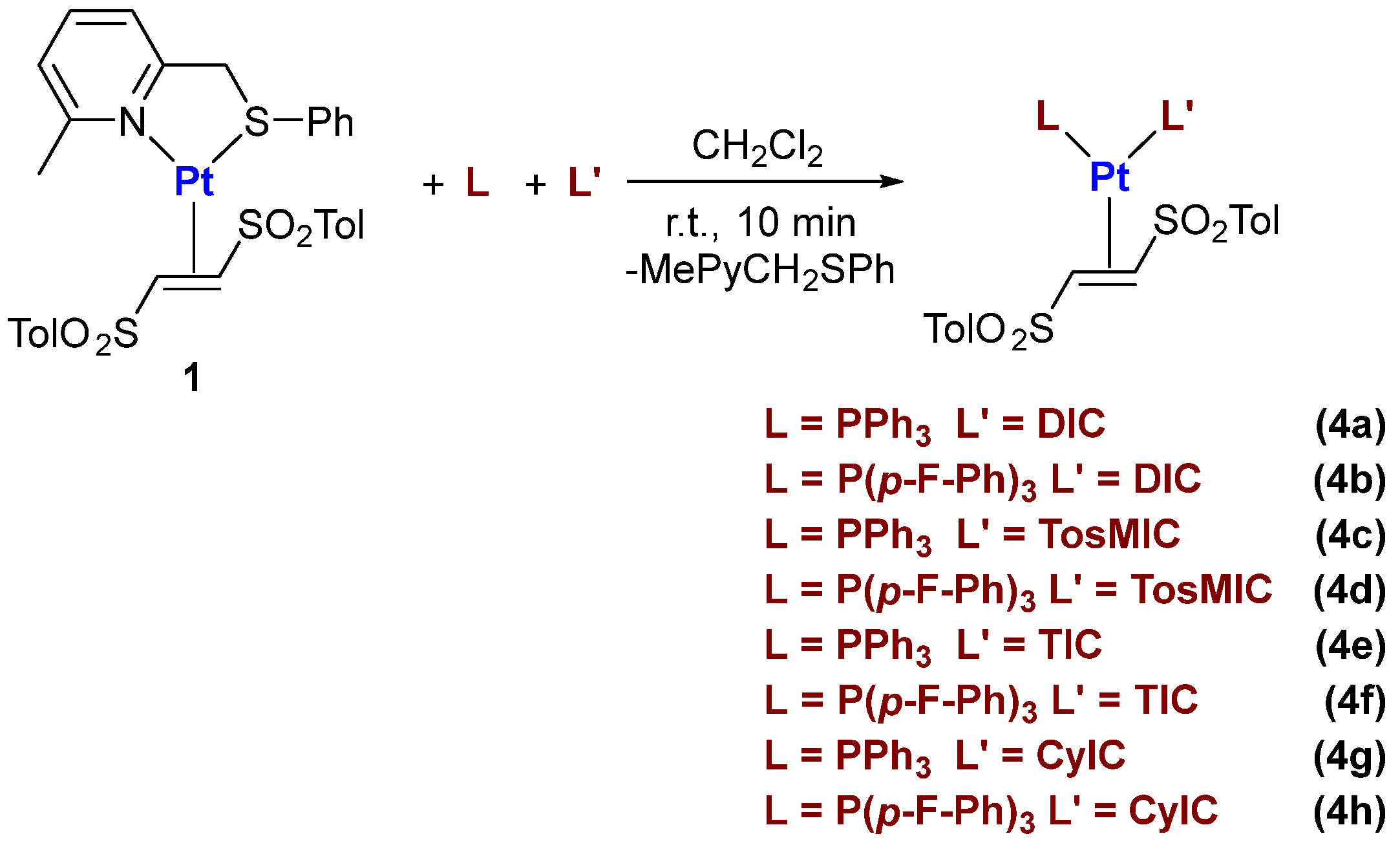
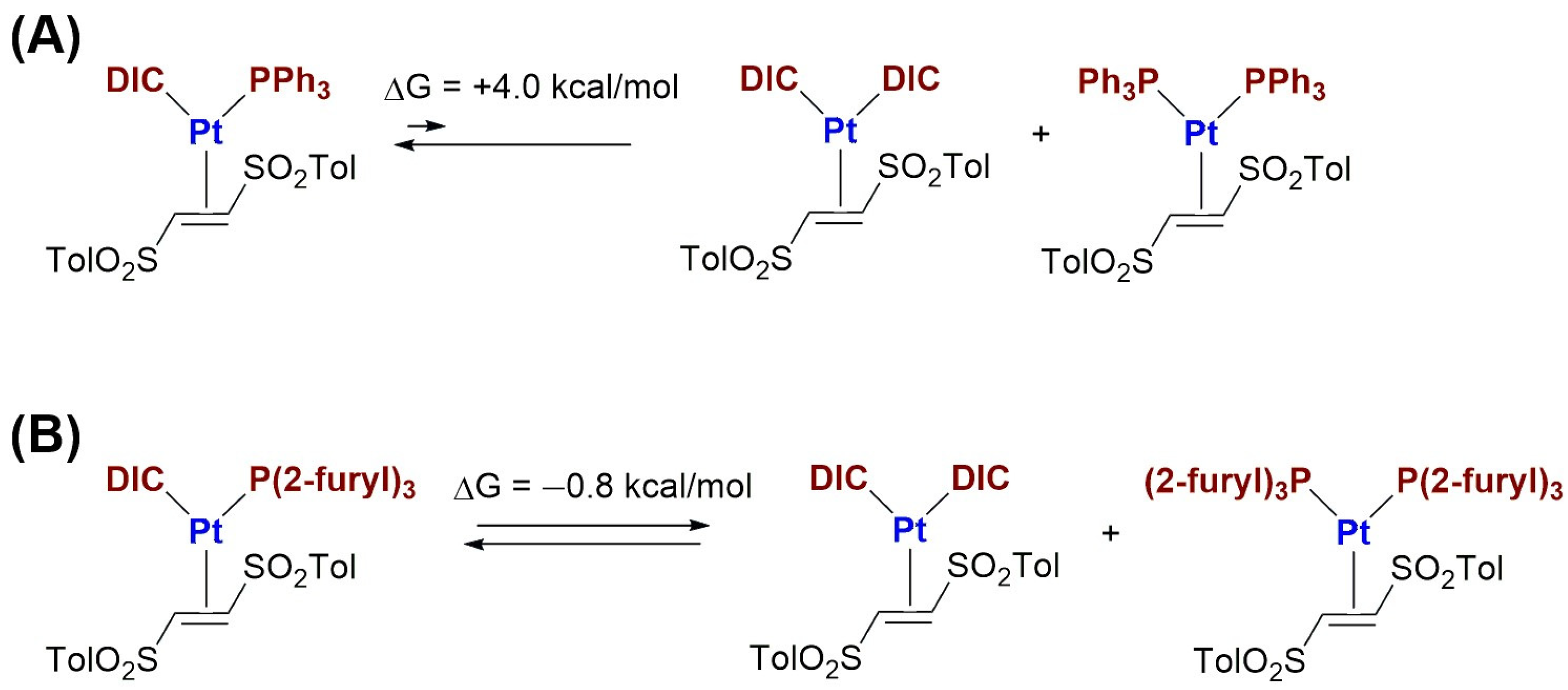
2.1. Synthesis and Characterization of Pt(0)-η2-(E)-1,2-Ditosylethene Complexes
2.2. Antiproliferative Activity of Pt(0)-η2-(E)-1,2-Ditosylethene Complexes towards Cancer Cells
3. Materials and Methods
3.1. Solvents and Reagents
3.2. Instruments
3.3. Synthesis of Complex 1
3.4. Synthesis of Pt(0) Complexes Bearing Isocyanide and/or Phosphine Ligands
3.5. Synthesis of Pt(0) Complexes Bearing N-Heterocyclic Carbene (NHC) Ligands
3.6. Computational Details
3.7. Cytotoxicity Assay
3.8. Crystal Structure Determination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rosenberg, B.; Vancamp, L.; Trosko, J.E.; Mansour, V.H. Platinum Compounds: A New Class of Potent Antitumour Agents. Nature 1969, 222, 385–386. [Google Scholar] [CrossRef]
- Lippert, B. Cisplatin. In Wiley eBooks; Verlag Helvetica Chimica Acta: Zürich, Switzerland, 1999. [Google Scholar] [CrossRef]
- Rixe, O.; Ortuzar, W.F.; Álvarez, M.; Parker, R.J.; Reed, E.; Paull, K.; Fojo, T. Oxaliplatin, Tetraplatin, Cisplatin, and Carboplatin: Spectrum of Activity in Drug-Resistant Cell Lines and in the Cell Lines of the National Cancer Institute’s Anticancer Drug Screen Panel. Biochem. Pharmacol. 1996, 52, 1855–1865. [Google Scholar] [CrossRef]
- Tsvetkova, D.; Ivanova, S. Application of Approved Cisplatin Derivatives in Combination Therapy against Different Cancer Diseases. Molecules 2022, 27, 2466. [Google Scholar] [CrossRef]
- De Biasi, A.R.; Villena-Vargas, J.; Adusumilli, P.S. Cisplatin-Induced Antitumor Immunomodulation: A Review of Preclinical and Clinical Evidence. Clin. Cancer Res. 2014, 20, 5384–5391. [Google Scholar] [CrossRef]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The Side Effects of Platinum-Based Chemotherapy Drugs: A Review for Chemists. Dalton Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef] [PubMed]
- Alassadi, S.; Pisani, M.J.; Wheate, N.J. A Chemical Perspective on the Clinical Use of Platinum-Based Anticancer Drugs. Dalton Trans. 2022, 51, 10835–10846. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Sanchez, A.; Bertrand, H. Pt(IV) Anticancer Prodrugs Bearing an Oxaliplatin Scaffold: What Do We Know about Their Bioactivity? Inorg. Chem. Front. 2024. [Google Scholar] [CrossRef]
- Choy, H.; Park, C.; Yao, M. Current Status and Future Prospects for Satraplatin, an Oral Platinum Analogue. Clin. Cancer Res. 2008, 14, 1633–1638. [Google Scholar] [CrossRef]
- Petruzzella, E.; Sirota, R.; Solazzo, I.; Gandin, V.; Gibson, D. Triple Action Pt(Iv) Derivatives of Cisplatin: A New Class of Potent Anticancer Agents That Overcome Resistance. Chem. Sci. 2018, 9, 4299–4307. [Google Scholar] [CrossRef] [PubMed]
- Scattolin, T.; Valente, G.; Luzietti, L.; Piva, M.; Demitri, N.; Lampronti, I.; Gambari, R.; Visentin, F. Synthesis and Anticancer Activity of Pt(0)-olefin Complexes Bearing 1,3,5-triaza-7-phosphaadamantane and N-heterocyclic Carbene Ligands. Appl. Organomet. Chem. 2021, 35, e6438. [Google Scholar] [CrossRef]
- Annunziata, A.; Cucciolito, M.E.; Imbimbo, P.; Silipo, A.; Ruffo, F. A Hydrophilic Olefin Pt(0) Complex Containing a Glucoconjugated 2-Iminopyridine Ligand: Synthesis, Characterization, Stereochemistry and Biological Activity. Inorganica Chim. Acta 2021, 516, 120092. [Google Scholar] [CrossRef]
- Uddin, J.; Dapprich, S.; Frenking, G.; Yates, B.F. Nature of the Metal−Alkene Bond in Platinum Complexes of Strained Olefins. Organometallics 1999, 18, 457–465. [Google Scholar] [CrossRef]
- Pryadun, R.S.; Gerlits, O.; Atwood, J.D. Structural Studies on Platinum Alkene Complexes and Precursors. J. Coord. Chem. 2006, 59, 85–100. [Google Scholar] [CrossRef]
- Clarke, M.L. Recent Advances in Homogeneous Catalysis Using Platinum Complexes. Polyhedron 2001, 20, 151–164. [Google Scholar] [CrossRef]
- Sprengers, J.W.; De Greef, M.; Duin, M.A.; Elsevier, C.J. Stable Platinum(0) Catalysts for Catalytic Hydrosilylation of Styrene and Synthesis of [Pt(Ar-bian)(H2-alkene)] Complexes. Eur. J. Inorg. Chem. 2003, 2003, 3811–3819. [Google Scholar] [CrossRef]
- Maliszewski, B.P.; Ritacco, I.; Beliš, M.; Hashim, I.I.; Tzouras, N.V.; Caporaso, L.; Cavallo, L.; Van Hecke, K.; Nahra, F.; Cazin, C.S.J.; et al. A Green Route to Platinum N-Heterocyclic Carbene Complexes: Mechanism and Expanded Scope. Dalton Trans. 2022, 51, 6204–6211. [Google Scholar] [CrossRef]
- Caliendo, C.; Fratoddi, I.; Russo, M.V. Sensitivity of a Platinum-Polyyne-Based Sensor to Low Relative Humidity and Chemical Vapors. Appl. Phys. Lett. 2002, 80, 4849–4851. [Google Scholar] [CrossRef]
- Scattolin, T.; Canovese, L.; Demitri, N.; Santo, C.; Visentin, F. The Importance of the Electronic and Steric Features of the Ancillary Ligands on the Rate of Cis–Trans Isomerization of Olefins Coordinated to Palladium(0) Centre. A Study Involving (Z)-1,2-Ditosylethene as Olefin Model. Polyhedron 2019, 173, 114144. [Google Scholar] [CrossRef]
- Scattolin, T.; Santo, C.; Demitri, N.; Canovese, L.; Visentin, F. Chemoselective Oxidative Addition of Vinyl Sulfones Mediated by Palladium Complexes Bearing Picolyl-N-Heterocyclic Carbene Ligands. Dalton Trans. 2020, 49, 5684–5694. [Google Scholar] [CrossRef] [PubMed]
- Scattolin, T.; Pangerc, N.; Lampronti, I.; Tupini, C.; Gambari, R.; Marvelli, L.; Rizzolio, F.; Demitri, N.; Canovese, L.; Visentin, F. Palladium (0) Olefin Complexes Bearing Purine-Based N-Heterocyclic Carbenes and 1,3,5-Triaza-7-Phosphaadamantane (PTA): Synthesis, Characterization and Antiproliferative Activity toward Human Ovarian Cancer Cell Lines. J. Organomet. Chem. 2019, 899, 120857. [Google Scholar] [CrossRef]
- Scattolin, T.; Logvinov, A.A.; Tzouras, N.V.; Cazin, C.S.J.; Nolan, S.P. Advances in the Synthesis and Applications of N-Heterocyclic Carbene Metal Complexes with a Focus on the Weak Base Route. Organometallics 2023, 42, 2692–2730. [Google Scholar] [CrossRef]
- Tsuchiya, K.; Kondo, H.; Nagashima, H. Ring Expansion of a Platinacyclopropane to a Platinacyclopentane by Double Insertion of Isocyanides into Pt−C Bonds. Organometallics 2007, 26, 1044–1051. [Google Scholar] [CrossRef]
- Moseley, K.; Maitlis, P.M. Acetylenes and Noble Metal Compounds. Part XI. Reactions of Di-Methyl Acetylenedicarboxylate with Dibenzylideneacetone–Palladium and –Platinum Complexes: Pallada- and Platina-Cyclopentadienes. J. Chem. Soc.-Dalton Trans. 1974, 169–175. [Google Scholar] [CrossRef]
- Canovese, L.; Santo, C.; Scattolin, T.; Visentin, F.; Bertolasi, V. Synthesis and Characterization of Palladacyclopentadiene Complexes with N-Heterocyclic Carbene Ligands. J. Organomet. Chem. 2015, 794, 288–300. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA Quantum Chemistry Program Package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef] [PubMed]
- Mardirossian, N.; Head-Gordon, M. Mapping the Genome of Meta-Generalized Gradient Approximation Density Functionals: The Search for B97M-V. J. Chem. Phys. 2015, 142, 074111. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Mardirossian, N.; Head-Gordon, M. ωB97M-V: A Combinatorially Optimized, Range-Separated Hybrid, Meta-GGA Density Functional with VV10 Nonlocal Correlation. J. Chem. Phys. 2016, 144, 214110. [Google Scholar] [CrossRef]
- Scattolin, T.; Pessotto, I.; Cavarzerani, E.; Canzonieri, V.; Orian, L.; Demitri, N.; Schmidt, C.; Casini, A.; Bortolamiol, E.; Visentin, F.; et al. Indenyl and Allyl Palladate Complexes BearingN-Heterocyclic Carbene Ligands: An Easily Accessible Class of New Anticancer Drug Candidates. Eur. J. Inorg. Chem. 2022, 2022, e202200103. [Google Scholar] [CrossRef]
- Scattolin, T.; Bortolamiol, E.; Caligiuri, I.; Rizzolio, F.; Demitri, N.; Visentin, F. Synthesis and Comparative Study of the Anticancer Activity of H3-Allyl Palladium(II) Complexes Bearing N-Heterocyclic Carbenes as Ancillary Ligands. Polyhedron 2020, 186, 114607. [Google Scholar] [CrossRef]
- Lausi, A.; Polentarutti, M.; Onesti, S.; Plaisier, J.R.; Busetto, E.; Bais, G.; Barba, L.; Cassetta, A.; Campi, G.; Lamba, D.; et al. Status of the Crystallography Beamlines at Elettra. Eur. Phys. J. Plus 2015, 130, 43. [Google Scholar] [CrossRef]
- Kabsch, W. Xds. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef]
- Agirre, J.; Atanasova, M.; Bagdonas, H.; Ballard, C.B.; Baslé, A.; Beilsten-Edmands, J.; Borges, R.J.; Brown, D.G.; Burgos-Mármol, J.J.; Berrisford, J.M. The CCP4 Suite: Integrative Software for Macromolecular Crystallography. Acta Crystallogr. Sect. D Struct. Biol. 2023, 79, 449–461. [Google Scholar] [CrossRef]
- Evans, P.R.; Murshudov, G.N. How Good Are My Data and What Is the Resolution? Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. checkCIF Validation ALERTS: What They Mean and How to Respond. Acta Crystallogr. Sect. E Crystallogr. Commun. 2020, 76, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and Development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An Update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Schrodinger LLC. The PyMOL Molecular Graphics System 2015. Available online: https://pymol.org/2/ (accessed on 17 February 2024).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
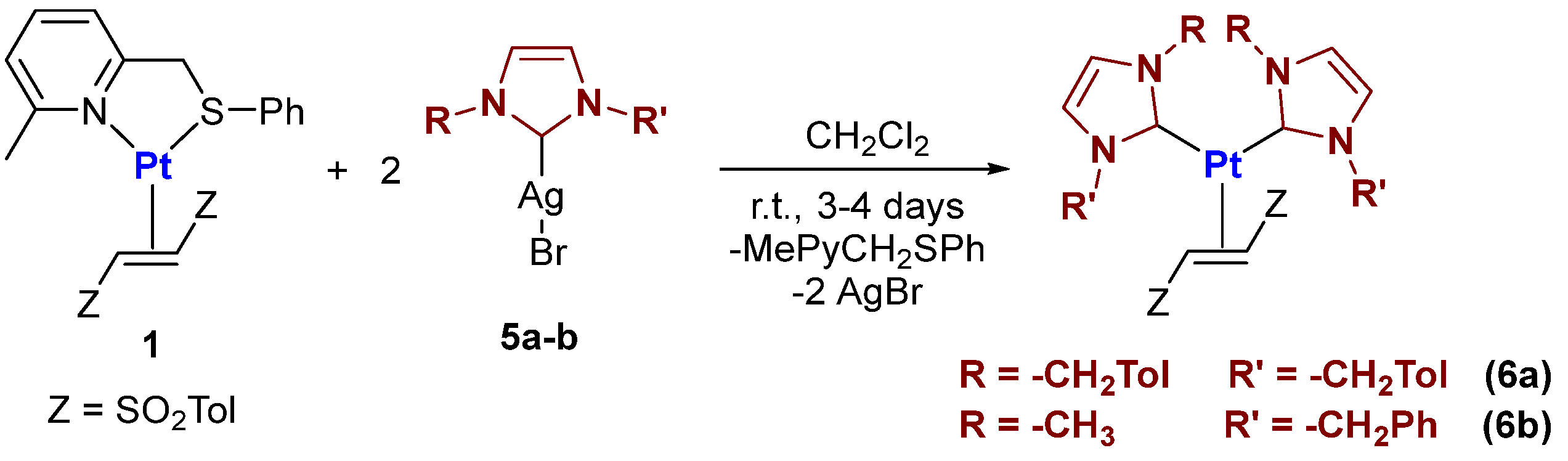{kind=link}
| Compound | IC50 (µM) | |||
|---|---|---|---|---|
| A2780 | A2780cis | MDA-MB-231 | MRC-5 | |
| Cisplatin | 0.19 ± 0.08 | 5.4 ± 0.1 | 6.4 ± 0.5 | 2.9 ± 0.1 |
| 2a | 5.5 ± 0.6 | >100 | 2.6 ± 0.4 | >100 |
| 2b | >100 | >100 | 36 ± 6 | >100 |
| 2c | 17.2 ± 0.8 | >100 | >100 | >100 |
| 3b | 5.4 ± 0.7 | 11 ± 6 | 5.52 ± 0.03 | 22 ± 6 |
| 3c | 2.6 ± 0.3 | 3.1 ± 0.9 | 3.4 ± 0.4 | 3.5 ± 0.1 |
| 3d | 1.18 ± 0.09 | 1.4 ± 0.2 | 1.2 ± 0.3 | 9 ± 1 |
| 4a | 35 ± 3 | >100 | 1.8 ± 0.6 | 59 ± 8 |
| 4b | 85 ± 23 | >100 | 30 ± 13 | >100 |
| 4c | 55 ± 17 | >100 | 1.0 ± 0.8 | 15 ± 3 |
| 4d | >100 | >100 | 0.4 ± 0.1 | 14 ± 1 |
| 4f | >100 | >100 | 21 ± 2 | >100 |
| 4h | 41 ± 10 | >100 | 37 ± 13 | >100 |
| 6a | 1.6 ± 0.1 | 10 ± 3 | 36 ± 11 | >100 |
| 6b | 1.9 ± 0.3 | 12.91 ± 0.06 | 5.4 ± 0.9 | 61 ± 13 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Compagno, N.; Piccolo, R.; Bortolamiol, E.; Demitri, N.; Rizzolio, F.; Visentin, F.; Scattolin, T. Platinum(0)-η2-1,2-(E)ditosylethene Complexes Bearing Phosphine, Isocyanide and N-Heterocyclic Carbene Ligands: Synthesis and Cytotoxicity towards Ovarian and Breast Cancer Cells. Molecules 2024, 29, 1119. https://doi.org/10.3390/molecules29051119
Compagno N, Piccolo R, Bortolamiol E, Demitri N, Rizzolio F, Visentin F, Scattolin T. Platinum(0)-η2-1,2-(E)ditosylethene Complexes Bearing Phosphine, Isocyanide and N-Heterocyclic Carbene Ligands: Synthesis and Cytotoxicity towards Ovarian and Breast Cancer Cells. Molecules. 2024; 29(5):1119. https://doi.org/10.3390/molecules29051119
Chicago/Turabian StyleCompagno, Nicola, Rachele Piccolo, Enrica Bortolamiol, Nicola Demitri, Flavio Rizzolio, Fabiano Visentin, and Thomas Scattolin. 2024. "Platinum(0)-η2-1,2-(E)ditosylethene Complexes Bearing Phosphine, Isocyanide and N-Heterocyclic Carbene Ligands: Synthesis and Cytotoxicity towards Ovarian and Breast Cancer Cells" Molecules 29, no. 5: 1119. https://doi.org/10.3390/molecules29051119
APA StyleCompagno, N., Piccolo, R., Bortolamiol, E., Demitri, N., Rizzolio, F., Visentin, F., & Scattolin, T. (2024). Platinum(0)-η2-1,2-(E)ditosylethene Complexes Bearing Phosphine, Isocyanide and N-Heterocyclic Carbene Ligands: Synthesis and Cytotoxicity towards Ovarian and Breast Cancer Cells. Molecules, 29(5), 1119. https://doi.org/10.3390/molecules29051119