
A Sustainable Improvement of ω-Bromoalkylphosphonates Synthesis to Access Novel KuQuinones
by
 , , , and
, , , and
Mattia Forchetta
1 ,
,
Valeria Conte
1,
Giulia Fiorani
2,
Pierluca Galloni
1,* and
Federica Sabuzi
1,* 1
Department of Chemical Science and Technologies, University of Rome Tor Vergata, Via della Ricerca Scientifica snc, 00133 Rome, Italy
2
Department of Molecular Sciences and Nanosystems, Ca’ Foscari University of Venezia, Via Torino 155, 30172 Venezia, Italy
*
Authors to whom correspondence should be addressed.
Organics 2021, 2(2), 107-117; https://doi.org/10.3390/org2020010
Submission received: 28 April 2021
/
Revised: 24 May 2021
/
Accepted: 1 June 2021
/
Published: 3 June 2021
(This article belongs to the Special Issue Feature Papers in Organics)
Abstract
:Owing to the attractiveness of organic phosphonic acids and esters in the pharmacological field and in the functionalization of conductive metal-oxides, the research of effective synthetic protocols is pivotal. Among the others, ω-bromoalkylphosphonates are gaining particular attention because they are useful building blocks for the tailored functionalization of complex organic molecules. Hence, in this work, the optimization of Michaelis–Arbuzov reaction conditions for ω-bromoalkylphosphonates has been performed, to improve process sustainability while maintaining good yields. Synthesized ω-bromoalkylphosphonates have been successfully adopted for the synthesis of new KuQuinone phosphonate esters and, by hydrolysis, phosphonic acid KuQuinone derivatives have been obtained for the first time. Considering the high affinity with metal-oxides, KuQuinones bearing phosphonic acid terminal groups are promising candidates for biomedical and photo(electro)chemical applications.
1. Introduction
Phosphonic acid (PA) functionalization of organic compounds is of great interest in various scientific areas. Several pharmacologically active compounds bearing a PA group (or the corresponding phosphonate salt) have been developed over the years [1,2]. Organic phosphonic acid derivatives, such as phosphonoformic or phosphonoacetic acids and many others, resulted in promising antiviral drugs [3,4] and, recently, the aromatic analogue, phosphonobenzoic acid, has been object of a theoretical study on Covid-SARS [5]. Additionally, fosmidomycin and its α-substituted derivatives are effective anti-malarian drugs [6], while fosfomycin is a widely used antibiotic [7]. Notably, the presence of a phosphonic acid group significantly improves organic molecules hydrophilicity, which is an essential feature for biomedical application [8].
PAs are also optimal hydrogen bond donors and acceptors; therefore, they are employed in supramolecular chemistry, for metal-organic frameworks and organic proton conductors [9,10,11,12]. The dianionic nature of the deprotonated form of PA is pivotal for covalently binding different metal oxides, in mono-, bi- or tri-dentate architectures [13]. In fact, PA is a robust anchoring group, widely adopted for organic molecules immobilization on metal oxide surfaces and nanoparticles [14,15,16,17,18,19,20,21]. Therefore, phosphonic acid functionalization of organic and metallorganic photosensitizers is a proficient tool to decorate metal oxides for their application in dye-sensitized solar cells, photoelectrochemical devices [22,23] as well as in organocatalysis [24,25,26,27].
With respect to photoelectrochemical applications, we have been recently involved in the use of metal-free quinoid compounds, i.e., KuQuinones (KuQs), as photosentisizers on indium-tin oxide (ITO) [28], NiO [29] and on SnO2 [30], due to their favorable electrochemical and photophysical properties [31]. In this respect, terminal hydroxy or carboxylic acid functionalities were introduced in KuQuinone structure to ensure a stable and effective metal oxide binding. Additionally, differently functionalized KuQs were tested as anti-proliferative agents against ovarian and colon cancer cells, with promising results [32,33].
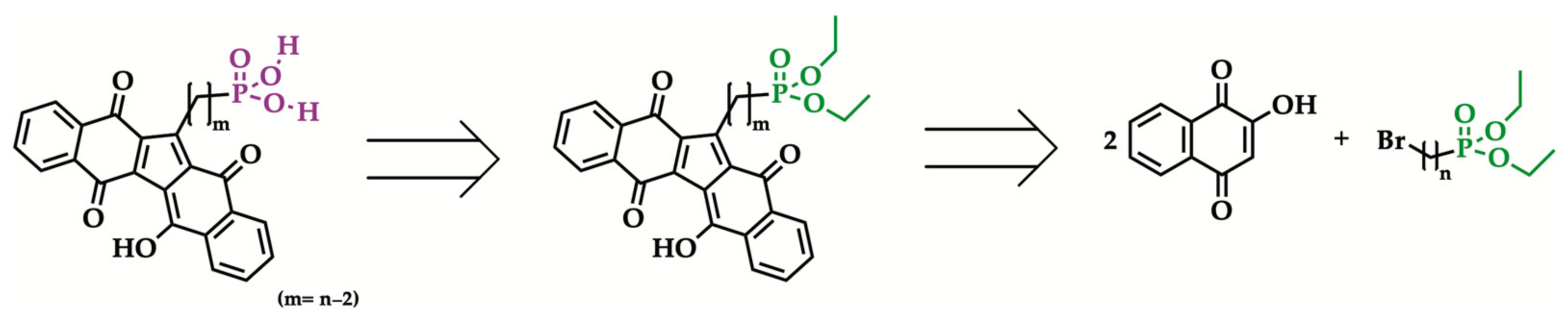
In an attempt to extend KuQuinones application in the photo(electro)chemical and biomedical fields, the synthesis of new KuQuinone phosphonic acid derivatives was investigated. As a matter of fact, compared to carboxylates, PAs are better anchoring groups in terms of stability [34,35], bond strength [36,37,38] and monolayer deposition reproducibility [39]. Additionally, PA functionalization is expected to enhance KuQ pharmacological applications. In this framework, according to the previously reported synthetic procedure, to obtain new KuQuinone phosphonic acid derivatives, the synthesis of suitable ω-bromoalkylphosphonate esters was required (Scheme 1) [32].
The first synthesis of alkylphosphonate esters has been proposed more than 100 years ago by Michaelis and Arbuzov [40,41]. Optimized reaction conditions required the use of an alkyl bromide with an excess of triethyl phosphite. Reactions proceeded solventless, at high temperatures, with good yields. Later, the synthesis of ω-bromoalkylphosphonates was also experienced with Arbuzov’s reaction [41]. Here, a dibromoalkane was used as the substrate. Nevertheless, such reactions lacked selectivity, because the di-substitution process occurred; therefore, a large excess of the dibromoalkane was required [42,43,44,45,46]. In that respect, although the authors claimed that the unreacted substrate can be recovered by fractional distillation and reused, it is worth mentioning that such a recovery procedure is actually problematic and time-consuming, because of the very similar boiling points of the reaction components. Therefore, in this paper, optimization of Michaelis–Arbuzov reaction conditions for the synthesis of ω-bromoalkylphosphonate esters is investigated, using an equimolar amount of reagents, to enhance process sustainability. The synthesized compounds are adopted for the synthesis of new KuQuinone derivatives.
2. Materials and Methods
All commercial reagents and solvents were purchased from Sigma Aldrich/MerckLife Science (KGaA, Darmstadt, Germany), with the highest degree of purity, and they were used without any further purification. UV-vis absorption spectra were recorded with a UV-Vis 2450 (Shimadzu, Kyoto, Japan) spectrophotometer. ATR-IR spectra were recorded with a FT-IR Nicolet iS50 (Thermo Scientific, Madison, WI, USA) spectrometer. Melting points were measured using a Büchi 512 melting point apparatus (Büchi, Flawil, CH) and all values were uncorrected. GC-MS analyses have been performed with a (Shimadzu, Kyoto, Japan) GCMS QP2010 Ultra system. 1H-NMR and 31P-NMR spectra were recorded with a Bruker AM400 NMR spectrometer (Bruker, Billerica, MA, USA) operating frequency of 400 MHz. Reactions with MW have been performed with a Microwave Accelerated Reaction System, Model MARS Xpress 5-CEM (CEM Corporation, Matthews, NC, USA). High-resolution mass spectra (HRMS) were recorded with a Bruker Compact QTOF instrument (Bruker, Billerica, MA, USA). Spectra have been acquired in positive or negative ion mode, with a mass resolution of R = 30,000.
2.1. Optimized Procedure for the Synthesis of Diethyl ω-Bromoalkylphosphonate
Glassware was flamed under nitrogen flow. The reaction flask was connected to a distillation apparatus, in order to distill the bromoethane produced during the process. A total of 75 mmol of α,ω-dibromoalkane were pre-heated at 140 °C; 75 mmol of triethyl phosphite were then added dropwise, within 2 h. The mixture was stirred for an additional hour and monitored by GC-MS. Pure diethyl ω-bromoalkylphosphonate was isolated by vacuum fractional distillation, with satisfactory yield. Product characterization has been performed with GC-MS, 1H and 31P NMR in CDCl3 (see Supplementary Materials). Reactions have been performed in duplicate and good reproducibility was observed.
Diethyl 6-bromohexylphosphonate (1). Colorless oil (9.19 g, 30.5 mmol, 40% yield). MS (EI, 70 eV): m/z = 221 [M − Br]+; m/z = 152 [•CH2P(=OH)(OCH2CH3)2]+; m/z = 125 [152-C2H3]+ [47,48]. 1H NMR (CDCl3, 400 MHz): δ 1.322 (t, J = 7.09 Hz, 6H), δ 1.366–1.509 (m, 4H), δ 1.562–1.667 (m, 4H), δ 1.819–1.908 (m, 2H), δ 3.402 (t, J = 6.74 Hz, 2H), δ 4.030–4.160 (m, 4H); 31P NMR (CDCl3, 400 MHz): δ 32.248 (s, 1P).
Diethyl 5-bromopentylphosphonate (2). Colorless oil (8.17 g, 29.5 mmol, 40% yield). MS (EI, 70 eV): m/z = 207 [M − Br]+; m/z = 152 [•CH2P(=OH)(OCH2CH3)2]+; m/z = 125 [152-C2H3]+ [47,48]. 1H NMR (CDCl3, 400 MHz): δ 1.322 (t, J = 7.06, 6H), δ 1.475–1.739 (m, 6H), δ 1.826–1.920 (m, 2H), δ 3.404 (t, J = 6.72 Hz, 2H), δ 4.013–4.166 (m, 4H); 31P NMR (CDCl3, 400 MHz): δ 31.926 (s, 1P).
Diethyl 4-bromobutylphosphonate (3). Colorless oil (4.24 g, 15.5 mmol, 20% yield). MS (EI, 70 eV): m/z = 193 [M − Br]+; m/z = 165 [193-C2H4]+; m/z = 137 [165-C2H4]+. 1H NMR (CDCl3, 400 MHz): δ 1.326 (t, J = 7.05, 6H), δ 1.730–1.821 (m, 4H), δ 1.918–2.002 (m, 2H), δ 3.409 (t, J = 6.56 Hz, 2H), δ 4.042–4.156 (m, 4H).; 31P NMR (CDCl3, 400 MHz): δ 31.400 (s, 1P).
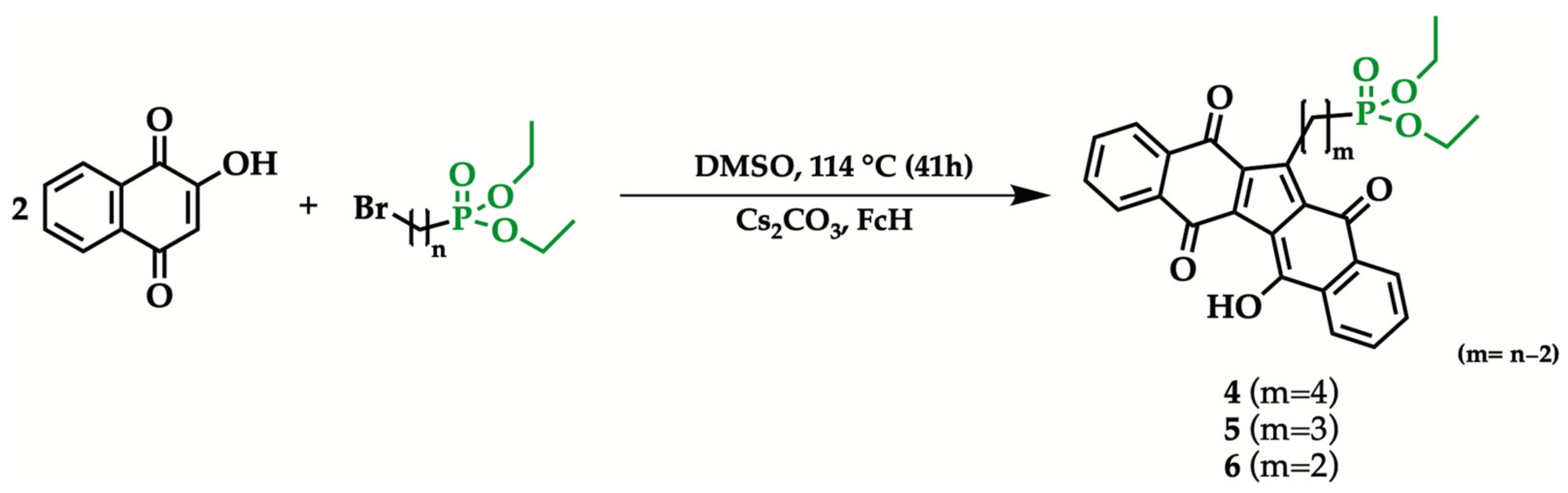
2.2. General Procedure for the Synthesis of 1-[n-(Diethyl phosphonyl) alkyl]KuQuinone
5.75 mmol of 2-hydroxy-1,4-naphthoquinone, 12 mmol of ω-bromoalkylphosphonate, 8 mmol of Cs2CO3, 0.33 mmol of sublimated ferrocene and 22 mL of dimethyl sulfoxide were mixed at 114 °C, for 41 h. The crude was then diluted with 100 mL of dichloromethane, filtered and extracted with NaCl saturated aqueous solution. The organic phase was subsequently dried over Na2SO4, filtered and concentrated. Purification was carried out by chromatography column (SiO2/CHCl3). The sample was dried and then further purified by precipitation with chloroform/hexane. The product was characterized by UV-vis and IR spectroscopy, HRMS, 1H, 13C and 31P NMR in CDCl3 (see Supplementary Materials).
1-[4-(Diethyl phosphonyl)butyl]KuQuinone (4). Purple powder (103.94 mg, 0.2 mmol, 7% yield) mp 185–188 °C. 1H NMR (CDCl3, 400 MHz): δ 1.303 (t, J = 7.08 Hz, 6H), δ 1.593–1.802 (m, 6H), δ 3.350–3.423 (t, J = 7.28 Hz, 2H), δ 4.026–4.147 (m, 4H), δ 7.622–7.729 (m, 4H), δ 8.102–8.199 (m, 4H), δ 18.072 (s, 1H). 13C NMR (CDCl3, 400 MHz): δ 181.052 and 177.973 (C=O); δ 136.089, 135.692, 135.127, 133.150, 131.905, 127,798, 127.341, 125.753 (aromatic carbons); δ 59.894, 27.354, 23.433, 22.360, 20.465, 16.428 (aliphatic carbons). 31P NMR (CDCl3, 400 MHz): δ 32.686 (s, 1P). UV−vis in CHCl3 [λmax, nm (ε, M−1cm−1)]: 569 (14,919); 532 (12,240). HRMS: m/z [M + Na]+ calculated for C29H27NaO7P 541.1387; found 541.1394.
1-[3-(Diethyl phosphonyl)propyl]KuQuinone (5). Purple powder (145.21 mg, 0.3 mmol, 10% yield) mp 204–206 °C. 1H NMR (CDCl3, 400 MHz): δ 1.281 (t, J = 7.08 Hz, 6H), δ 1.862–2.053 (m, 4H), δ 3.486 (t, J = 7.09, 2H), δ 3.991–4.152 (m, 4H), δ 7.628–7.780 (m, 4H), δ 8.120–8.238 (m, 4H), δ 18.150 (s, 1H). 13C NMR (CDCl3, 400 MHz): δ 180.876 and 178.090 (C=O); δ 135.664, 135.133, 133.125, 132.638, 131.836, 127,827, 127.344, 125.515 (aromatic carbons); δ 61.437, 26.312, 24.920, 22.263, 16.524 (aliphatic carbons). 31P NMR (CDCl3, 400 MHz): δ 32.337 (s, 1P). UV−vis in CHCl3 [λmax, nm (ε, M−1cm−1)]: 567 (14,851); 529 (11,351). HRMS: m/z [M + Na]+ calculated for C28H25NaO7P 527.1230; found 527.1234.
1-[2-(Diethyl phosphonyl)ethyl]KuQuinone (6). Purple powder (152.60 mg, 0.3 mmol, 11% yield) mp 220–222 °C. 1H NMR (CDCl3, 400 MHz): δ 1.423 (t, J = 7.00 Hz, 6H), δ 2.102–2.224 (m, 2H), δ 3.602–3.702 (m, 2H), δ 4.157–4.320 (m, 4H), δ 7.645–7.764 (m, 4H), δ 8.163–8.228 (m, 4H), δ 18.147 (s, 1H). 13C NMR (CDCl3, 400 MHz): δ 30.835 (s, 1P). δ 180.888 and 178.309 (C=O); δ 135.613, 135.183, 133.164, 132.404, 131.861, 127,925, 127.363, 125.434 (aromatic carbons); δ 61.827, 24.772, 20.637, 16.621 (aliphatic carbons). 31P NMR (CDCl3, 400 MHz). UV−vis in CHCl3 [λmax, nm (ε, M−1cm−1)]: 563 (14,544); 526 (11,367). HRMS: m/z [M + Na]+ calculated for C27H23NaO7P 513.1074; found 513.1069.
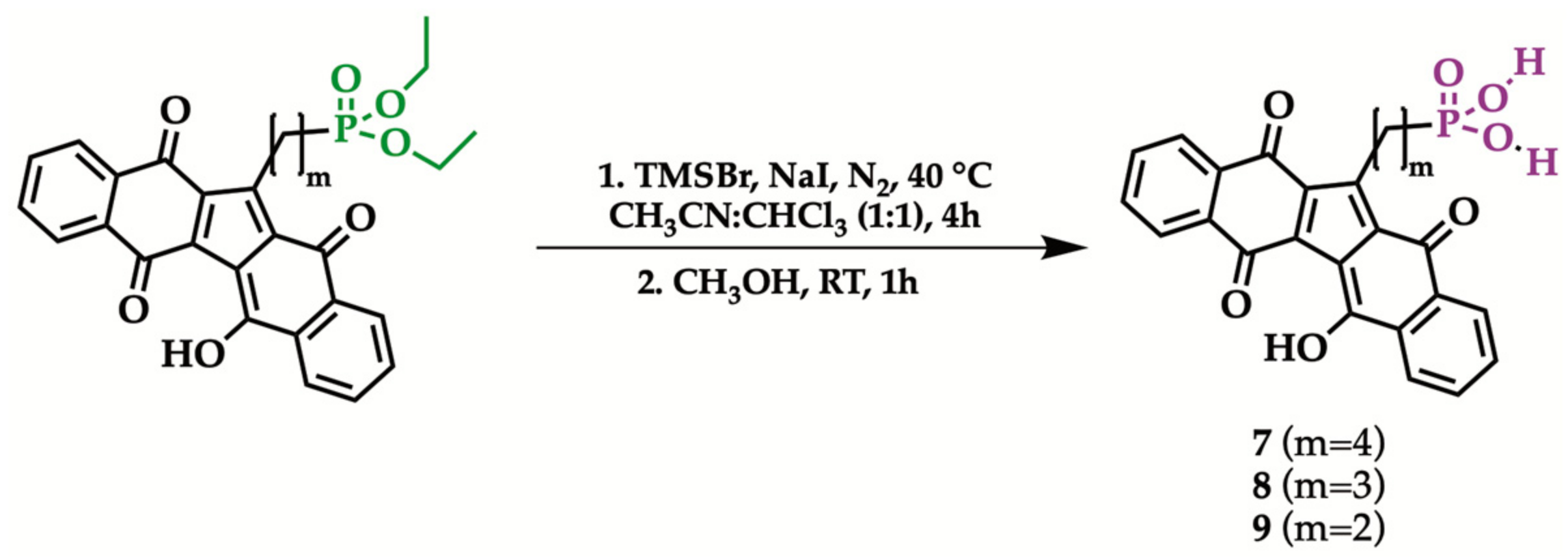
2.3. Hydrolysis of Diethyl Phosphonate KuQuinone Derivatives
Glassware was flamed under nitrogen flow before use. A total of 0.1 mmol of diethyl phosphonate KuQuinone and 5 mmol of NaI were mixed in 16 mL of dry acetonitrile: chloroform solution (1:1 v/v) until a deep purple solution was observed. Subsequently, 5 mmol of bromotrimethylsilane were added and the mixture was stirred for 4 h, at 40 °C, under an inert atmosphere. The crude was then diluted with 15 mL of chloroform, filtered and concentrated under vacuum, obtaining the bis-(trimethylsilyl)phosphonate intermediate, as a brownish oil. The silyl ester was converted to the corresponding acid by hydrolysis with 30 mL of methanol, under magnetic stirring, within an hour. Phosphonic acid, in form of a purple precipitate, was isolated by vacuum filtration, washed with diethyl ether and characterized by 1H and 31P NMR in DMSO-d6, IR and HRMS (see Supplementary Materials).
1-[4-(Dihydroxyphosphonyl)butyl]KuQuinone (7). Purple powder (44.83 mg, 0.097 mmol, 97% yield) mp > 250 °C. 1H NMR (DMSO-d6, 400 MHz): δ 1.493–1.617 (m, 6H), δ 7.529–7.697 (m, 4H), δ 7.891–8.035 (m, 4H). 31P NMR (DMSO-d6, 400 MHz): δ 26.653 (s, 1P). HRMS: m/z [M − H]− calculated for C25H18O7P 461.0796; found 461.0786.
1-[3-(Dihydroxyphosphonyl)propyl]KuQuinone (8). Purple powder (42.56 mg, 0.095 mmol, 95% yield) mp > 250 °C. 1H NMR (DMSO-d6, 400 MHz): δ 1.493–1.617 (m, 6H), δ 7.529–7.697 (m, 4H), δ 7.891–8.035 (m, 4H). 31P NMR (DMSO-d6, 400 MHz): δ 26.534 (s, 1P). HRMS: m/z [M − H]− calculated for C24H16O7P 447.0639; found 447.0625.
1-[2-(Dihydroxyphosphonyl)ethyl]KuQuinone (9). Purple powder (43.38 mg, 0.1 mmol, 100% yield) mp > 250 °C. 1H NMR (DMSO-d6, 400 MHz): δ 1.692–1.847 (m, 2H), δ 7.509–7.677 (m, 4H), δ 7.921–8.045 (m, 4H). 31P NMR (DMSO-d6, 400 MHz): δ 26.483 (s, 1P). HRMS: m/z [M - H]− calculated for C23H14O7P 433.0483; found 433.0478.
3. Results and Discussion
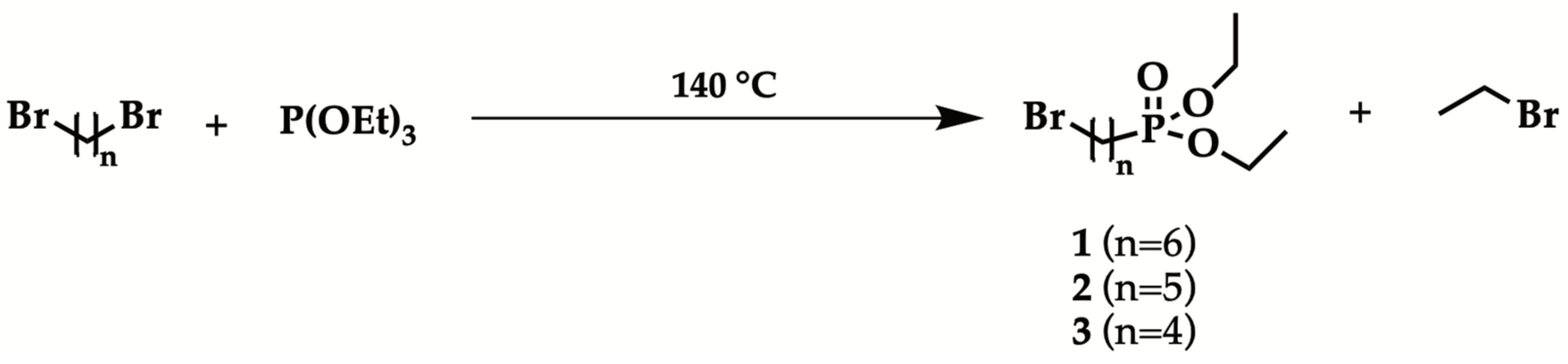
Synthesis of diethyl ω-bromoalkylphosphonates has been explored, in order to optimize Arbuzov reaction conditions [41]. Previous works on phosphonate esters mono-functionalization of dibromoalkanes highlighted that a high excess (from 3 to 20 equivalents) of substrate was required to avoid di-substitution reaction [42,43,44,45,46]. In this study, 1,4-dibromobutane, 1,5-dibromopentane and 1,6-dibromohexane have been selected as substrates to perform Arbuzov synthesis. The reactions have been carried out solventless, at 140 °C, using equimolar amount of triethyl phosphite with respect to the substrate, in order to improve process sustainability, thus reducing waste (Scheme 2). The reactions were monitored by GC-MS. Products isolation has been performed by fractional distillation under vacuum. The results are reported in Table 1.
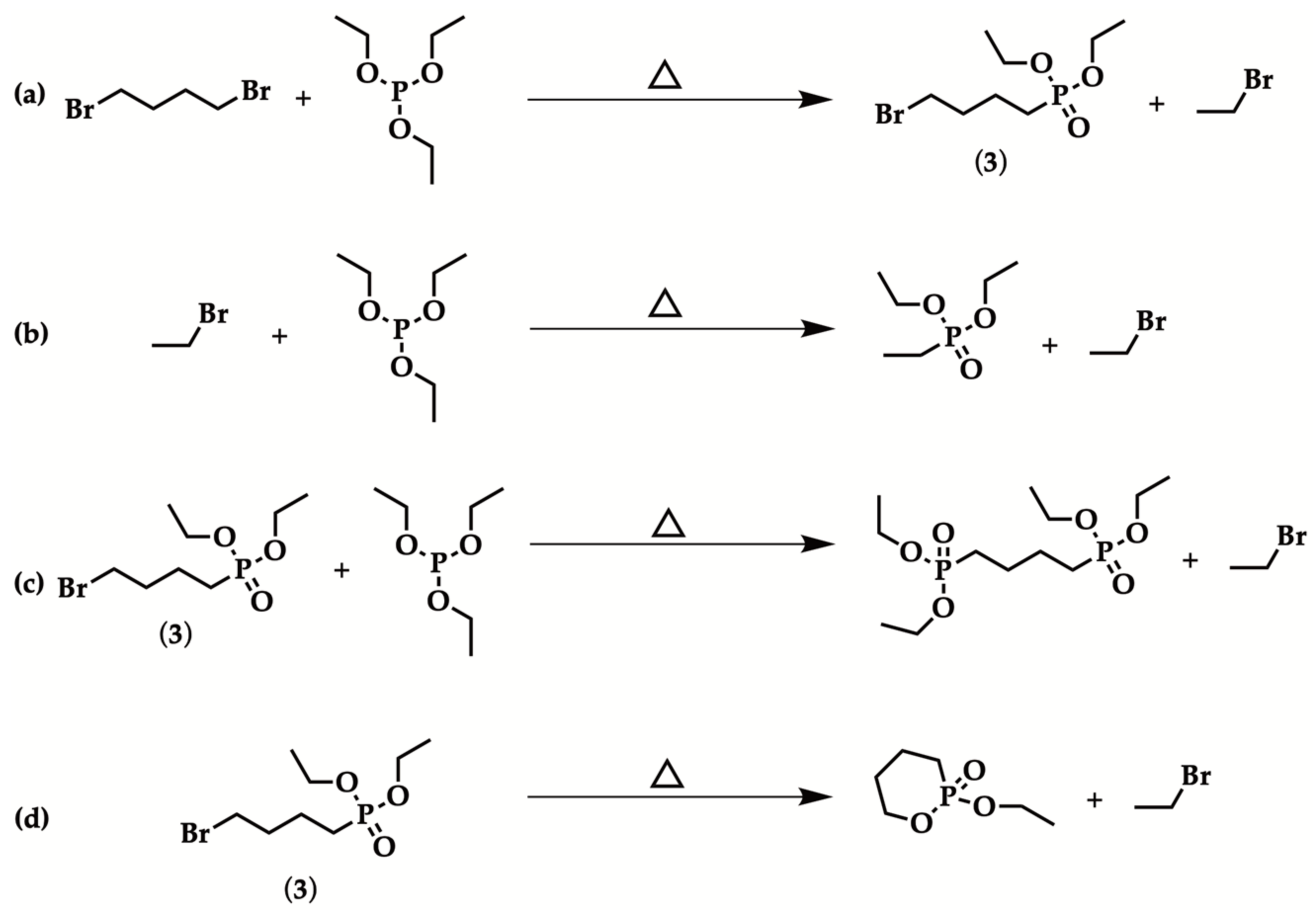
Solvent-free Arbuzov reaction between triethyl phosphite and 1,4-dibromobutane, in equimolar amount, was performed following a previously reported procedure (Table 1, Entry 1) [49,50]. However, in the adopted conditions, GC-MS analysis revealed that diethyl ethylphosphonate was the main product. As a matter of fact, during the Arbuzov reaction, bromoethane is formed as a byproduct (Scheme 3a) and it readily reacted with triethyl phosphite, leading to diethyl ethylphosphonate, in a high amount (Scheme 3b). Only traces of diethyl 4-bromobutylphosphonate (3) and the di-substituted product were detected.
To improve the experimental conditions, the same reaction was performed while distilling the generated bromoethane (Table 1, Entry 2); this was also useful to monitor the progress of the reaction. Product analysis pointed out that di-substitution reaction (Scheme 3c) and intramolecular cyclization of 3 (Scheme 3d) occurred, thus forming a stable six-membered heterocycle [44]. Additionally, in this case, only traces of the mono-substituted product were detected. Microwave irradiation was attempted [51,52], using equimolar reactants ratio, and an irradiation power of 150 W for 5 min; however, the reaction lacked in selectivity (Table 1, Entry 3).
In order to avoid cyclization reaction to a stable 6-membered ring, 1,6-dibromohexane was used as the substrate, experimenting with the synthesis of diethyl 6-bromohexylphosphonate (1). Moreover, to promote mono-substitution reaction, triethyl phosphite was added dropwise in 30 minutes [44]. However, in such conditions, di-phosphonation process was again the predominant one, while 1 was isolated in very low yield (Table 1, Entry 4). It is noteworthy that slower triethyl phosphite addition (in about two hours) allowed to achieve 1, with high selectivity and satisfactory yield (40%, Table 1 Entry 5). Thus, such an optimized procedure was employed to synthesize diethyl 5-bromopentylphosphonate (2) (40% yield, Table 1, Entry 6) and 3 (20% yield, Table 1, Entry 7), in acceptable yields. Nevertheless, in the case of 3, it was necessary to further purify the product, since the intramolecular cyclization side-reaction occurred (Scheme 3d).
The obtained diethyl ω-bromoalkylphosphonates (1–3) were then employed as reagents to synthesize new KuQuinone derivatives (4–6), bearing a diethyl phosphonate terminal group. KuQ synthesis was executed following the previously reported one-pot procedure [32], in which 2-hydroxy-1,4-naphthoquinone is reacted with an excess of an appropriate alkyl bromide, in the presence of a base and ferrocene (FcH) in catalytic amount (Scheme 4). Overall, three KuQuinone phosphonate derivatives, differing in side-chain length, were synthesized with yields (7 to 11%, Table 2) that compare well with that of other KuQuinone analogues [29,30,33].
To achieve KuQuinone phosphonic acids, KuQ-phosphonate esters hydrolysis was performed [53]. Reaction with 48% hydrobromic acid, at reflux, was first studied [54]. Despite the harsh conditions adopted, the phosphonic acid was not obtained. Acid hydrolysis was then performed with 0.5 M of hydrochloric acid, under microwave irradiation, with no results, likely because of the poor KuQuinone solubility in water [55]. Additionally, hydrolysis with bromotrimethylsilane (TMSBr) [56,57,58] was not successful. At this point, considering a seminal work by Morita [59], KuQuinone phosphonate ester hydrolysis has been performed with TMSBr in the presence of NaI (Table 3). In fact, because of the higher nucleophilicity of the iodide compared to other halides, trimethylsilyl intermediate formation is favored. Therefore, the reaction was carried out, dissolving 0.1 mmol of KuQ phosphonate ester in dry chloroform/acetonitrile (1:1), with a large excess of NaI and TMSBr (5 mmol), under N2 atmosphere, at 40 °C. After 4 h, the generated intermediate (trimethylsililphosphonate ester) was converted to the corresponding acid by methanolysis at room temperature (Scheme 5). The reaction was checked with TLC and interrupted when the ester band disappeared. In these conditions, almost quantitative conversions of KuQuinone phosphonate esters 4–6 to phosphonic acids 7–9 were achieved.
Note that KuQ phosphonate esters were highly soluble in all conventional organic solvents. Conversely, phosphonic acid derivatives were insoluble in apolar organic solvents and they showed only poor solubility in methanol and dimethyl sulfoxide. Solubility was significantly enhanced in water in the presence of a base, which allows phosphonic acid deprotonation and dissolution [60].
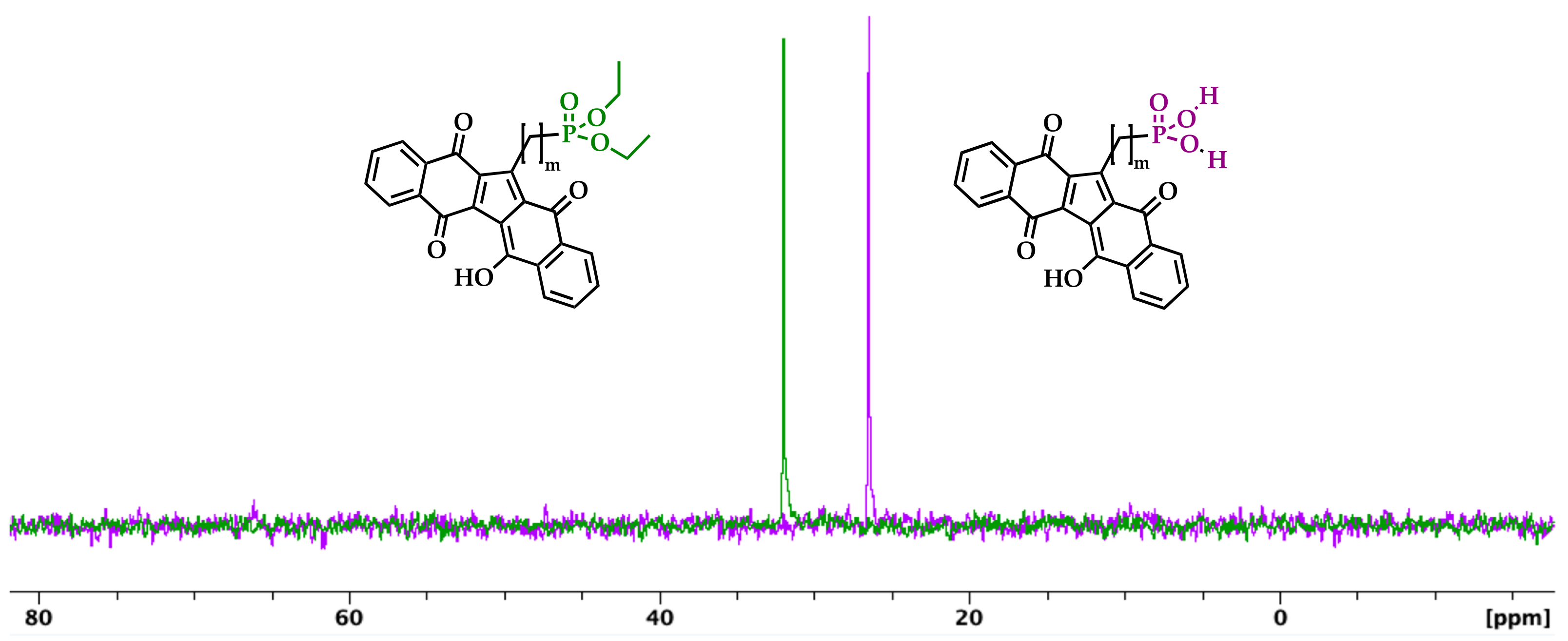
Therefore, KuQ phosphonic acid characterization was performed with NMR in DMSO-d6. 1H NMR spectra were characterized by a large and intense peak at about 3.4 ppm, which is typical of organic phosphonic acids (Figures S13, S17 and S21), being attributable to the hydroxyl groups of PA moiety, as well as to water traces [61]. Such intense peak overlaps with some KuQ aliphatic signals in the 1H NMR spectrum of the products, that are also broad and not well-resolved, likely because of the compound’s low solubility in DMSO. Conversely, KuQuinones characteristic peaks were clearly observed in the aromatic region of the spectra. On the other hand, 31P NMR analysis unambiguously confirmed the effectiveness of the hydrolysis [62]. In fact, by comparing the 31P NMR spectra in DMSO-d6 of the phosphonate esters with the corresponding phosphonic acids (Figure 1), a shift of about 5 ppm fields was observed, according to literature data [62].
4. Conclusions
In this paper, Arbuzov reaction for the synthesis of ω-bromoalkylphosphonates has been optimized. In fact, the main drawback in published synthetic procedures of ω-bromoalkylphosphonates was the large excess of the substrate, i.e., a α,ω-dibromoalkane, in order to prevent the di-substitution side process. Here, we report that slow dropwise addition of triethyl phosphite to 1 equivalent a α,ω-dibromoalkane leads to the desired product, with satisfactory yields. Importantly, reactions occur at 140 °C, while distilling bromoethane, that is formed as the first byproduct. Note that reactions performed using 1,4-dibromobutane as the substrate lead to a high amount of the cyclization side-product, thus lowering yields.
The optimized reaction conditions allow to significantly reduce the amount of the substrate, consequently reducing α,ω-dibromoalkane waste. Moreover, substrate scope can be further extended to obtain a library of ω-bromoalkylphosphonates.
The synthesized ω-bromoalkylphosphonates have been experimented in the synthesis of KuQuinone diethyl phosphonate derivatives, following a previously reported procedure. Hence, novel KuQuinone phosphonate esters have been synthesized and their hydrolysis with TMSBr and NaI allowed to access new KuQuinone phosphonic acids, with almost quantitative yields.
The introduction of a phosphonate ester in KuQ side chain sensibly enhances the solubility of such compounds in organic solvents. More importantly, a phosphonic acid functionality is strategic to exploit KuQuinones in different applications, such as in photo(electro)chemical devices anchored on suitable metal oxide nanoparticles, as well as in photocatalysis and biomedical applications.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/org2020010/s1, Figure S1: 1H NMR spectrum of 1 in CDCl3; Figure S2: 31P NMR spectrum of 1 in CDCl3; Figure S3: Mass spectrum of 1; Figure S4: 1H NMR spectrum of 2 in CDCl3; Figure S5: 31P NMR spectrum of 2 in CDCl3; Figure S6: Mass spectrum of 2; Figure S7: 1H NMR spectrum of 3 in CDCl3; Figure S8: 31P NMR spectrum of 3 in CDCl3; Figure S9: Mass spectrum of 3; Figure S10: 1H NMR spectrum of 4 in CDCl3; Figure S11: 13C NMR spectrum of 4 in CDCl3; Figure S12: 31P NMR spectrum of 4 in CDCl3; Figure S13: HRMS spectrum of 4; Figure S14: UV-vis spectrum of 4; Figure S15: ATR-IR spectrum of 4; Figure S16: 1H NMR spectrum of 7 in DMSO-d6; Figure S17: 31P NMR spectrum of 7 in DMSO-d6; Figure S18: HRMS of 7; Figure S19: ATR-IR spectrum of 7; Figure S20: 1H NMR spectrum of 5 in CDCl3; Figure S21: 13C NMR spectrum of 5 in CDCl3; Figure S22: 31P NMR spectrum of 5 in CDCl3; Figure S23: HRMS spectrum of 5; Figure S24: UV-vis spectrum of 5 in CHCl3; Figure S25: ATR-IR spectrum of 5; Figure S26: 1H NMR spectrum of 8 in DMSO-d6; Figure S27: 31P NMR spectrum of 8 in DMSO-d6; Figure S28: HRMS spectrum of 8; Figure S29: ATR-IR spectrum of 8; Figure S30: 1H NMR spectrum of 6 in CDCl3; Figure S31: 13C NMR spectrum of 6 in CDCl3; Figure S32: 31P NMR spectrum of 6 in CDCl3; Figure S33: HRMS spectrum of 6; Figure S34: UV-vis spectrum of 6 in CHCl3; Figure S35: ATR-IR spectrum of 6; Figure S36: 1H NMR spectrum of 9 in DMSO-d6; Figure S37: 31P NMR spectrum of 9 in DMSO-d6; Figure S38: HRMS spectrum of 9; Figure S39: ATR-IR spectrum of 9.
Author Contributions
Conceptualization, F.S. and P.G.; methodology, M.F. and P.G.; validation, M.F., F.S. and P.G.; formal analysis, M.F. and G.F.; investigation, M.F., F.S. and P.G.; resources, P.G.; data curation, M.F. and F.S.; writing—original draft preparation, M.F. and F.S.; writing—review and editing, F.S., P.G. and V.C.; supervision, P.G. and V.C.; project administration, F.S. and P.G.; funding acquisition, F.S. and P.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by University of Rome Tor Vergata, grant HYPHOTOCAT project, “Beyond borders”.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
YERUN Network is acknowledged for the opportunity to start this project. Claudia Mazzuca is gratefully acknowledged for ATR-IR measurements.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lyagin, I.; Efremenko, E. Enzymes, Reacting with Organophosphorus Compounds as Detoxifiers: Diversity and Functions. Int. J. Mol. Sci. 2021, 22, 1761. [Google Scholar] [CrossRef]
- Wiemer, A.J.; Wiemer, D.F. Prodrugs of phosphonates and phosphates: Crossing the membrane barrier. Top. Curr. Chem. 2015, 360, 115–160. [Google Scholar] [CrossRef] [Green Version]
- Tan, E.L.; Ooi, E.E.; Lin, C.Y.; Tan, H.C.; Ling, A.E.; Lim, B.; Stanton, L.W. Inhibition of SARS coronavirus infection in vitro with clinically approved antiviral drugs. Emerg. Infect. Dis. 2004, 10, 581–586. [Google Scholar] [CrossRef] [Green Version]
- De Clercq, E. Clinical Potential of the Acyclic Nucleoside Phosphonates Cidofovir, Adefovir, and Tenofovir in Treatment of DNA Virus and Retrovirus Infections. E. Clin. Microbiol. Rev. 2003, 16, 569–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, W. Covid-19 Drug Design via Quantum Mechanical Principles Leads to a New Corona-SARS Anti- Viral Candidate 2-Phosphono-Benzoic-Acid. J. Theor. Comput. Sci. 2020. [Google Scholar] [CrossRef]
- Verbrugghen, T.; Vandurm, P.; Pouyez, J.; Maes, L.; Wouters, J.; Van Calenbergh, S.J. Alpha-Heteroatom Derivatized Analogues of 3-(Acetylhydroxyamino)propyl Phosphonic Acid (FR900098) as Antimalarials. Med. Chem. 2013, 56, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Falagas, M.E.; Vouloumanou, E.K.; Samonis, G.; Vardakas, K.Z. Fosfomycin. Clin. Microbiol. Rev. 2016, 29, 321–347. [Google Scholar] [CrossRef] [Green Version]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: Importance and enhancement techniques. ISRN Pharm. 2012, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, X.; Hu, J.Y.; Luo, J.; Liao, W.M.; He, J. Conductive Metal–Organic Frameworks: Mechanisms, Design Strategies and Recent Advances. Top. Curr. Chem. 2020, 378, 27. [Google Scholar] [CrossRef]
- Hix, G.B.; Caignaert, V.; Rueff, J.M.; Le Pluart, L.; Warren, J.E.; Jaffrès, P.A. A Supramolecular Ladderlike Structure Formed by the Auto-Assembly of Benzene-1,3,5-triphosphonic Acid. Cryst. Growth Des. 2007, 7, 208–211. [Google Scholar] [CrossRef]
- Sopkova-de Oliveira Santos, J.; Montouillout, V.; Fayon, F.; Fernandez, C.; Delain-Bioton, L.; Villemin, D.; Jaffrès, P.A. Assembly of benzene-1,3,5-tris(methylenephosphonic acid) and guanidinium salt: Single crystal-X-ray characterisation and 31P solid state NMR investigations. New J. Chem. 2004, 28, 1244–1249. [Google Scholar] [CrossRef]
- Jiménez-García, L.; Kaltbeitzel, A.; Enkelmann, V.; Gutmann, J.S.; Klapper, M.; Müllen, K. Organic Proton-Conducting Molecules as Solid-State Separator Materials for Fuel Cell Applications. Adv. Funct. Mater. 2011, 21, 2216–2224. [Google Scholar] [CrossRef]
- Hotchkiss, P.J.; Jones, S.C.; Paniagua, S.A.; Sharma, A.; Kippelen, B.; Armstrong, N.R.; Marder, S.R. The Modification of Indium Tin Oxide with Phosphonic Acids: Mechanism of Binding, Tuning of Surface Properties, and Potential for Use in Organic Electronic Applications. Acc. Chem. Res. 2012, 45, 337–346. [Google Scholar] [CrossRef]
- Queffélec, C.; Petit, M.; Janvier, P.; Knight, D.A.; Bujoli, B. Surface Modification Using Phosphonic Acids and Esters. Chem. Rev. 2012, 112, 3777–3807. [Google Scholar] [CrossRef]
- Pujari, S.P.; Scheres, L.; Marcelis, A.T.M.; Zuilhof, H. Covalent Surface Modification of Oxide Surfaces. Angew. Chem. Int. Ed. 2014, 53, 6322–6356. [Google Scholar] [CrossRef]
- Boissezon, R.; Muller, J.; Beaugeard, V.; Monge, S.; Robin, J.J. Organophosphonates as anchoring agents onto metal oxide-based materials: Synthesis and applications. RSC Adv. 2014, 4, 35690–35707. [Google Scholar] [CrossRef]
- Villemin, D.; Moreau, B.; Siméon, F.; Maheut, G.; Fernandez, C.; Montouillout, V.; Caignaert, V.; Jaffrès, P.A. A one step process for grafting organic pendants on alumina via the reaction of alumina and phosphonate under microwave irradiation. Chem. Commun. 2001, 2060–2061. [Google Scholar] [CrossRef]
- Zeininger, L.; Portilla, L.; Halik, M.; Hirsch, A. Quantitative Determination and Comparison of the Surface Binding of Phosphonic Acid, Carboxylic Acid, and Catechol Ligands on TiO2 Nanoparticles. Chem. Eur. J. 2016, 22, 13506–13512. [Google Scholar] [CrossRef] [PubMed]
- Lafond, V.; Gervais, C.; Maquet, J.; Prochnow, D.; Babonneau, F.; Mutin, P.H. 17O MAS NMR Study of the Bonding Mode of Phosphonate Coupling Molecules in a Titanium Oxo-Alkoxo-Phosphonate and in Titania-Based Hybrid Materials. Chem. Mater. 2003, 15, 4098–4103. [Google Scholar] [CrossRef]
- Holland, G.P.; Sharma, R.; Agola, J.O.; Amin, S.; Solomon, V.C.; Singh, P.; Buttry, D.A.; Yarger, J.L. NMR Characterization of Phosphonic Acid Capped SnO2 Nanoparticles. Chem. Mater. 2007, 19, 2519–2526. [Google Scholar] [CrossRef]
- Tudisco, C.; Oliveri, V.; Cantarella, M.; Vecchio, G.; Condorelli, G.G. Cyclodextrin Anchoring on Magnetic Fe3O4 Nanoparticles Modified with Phosphonic Linkers. Eur. J. Inorg. Chem. 2012, 32, 5323–5331. [Google Scholar] [CrossRef]
- Zhang, L.; Cole, J.M. Anchoring Groups for Dye-Sensitized Solar Cells. ACS Appl. Mater. Interfaces 2015, 7, 3427–3455. [Google Scholar] [CrossRef]
- Büttner, A.; Brauchli, S.Y.; Vogt, R.; Constable, E.C.; Housecroft, C.E. Combining phosphonic acid-functionalized anchoring ligands with asymmetric ancillary ligands in bis(diimine)copper(I) dyes for dye sensitized solar cells. RSC Adv. 2016, 6, 5205–5213. [Google Scholar] [CrossRef] [Green Version]
- De Tovar, J.; Romero, N.; Denisov, S.A.; Bofill, R.; Gimbert-Surinach, C.; Ciuculescu-Pradines, D.; Drouet, S.; Llobet, A.; Lecante, P.; Colliere, V.; et al. Light-driven water oxidation using hybrid photosensitizer-decorated Co3O4 nanoparticles. Mat. Today Energy 2018, 9, 506–515. [Google Scholar] [CrossRef]
- Xie, G.; Feng, D.; Ma, X. 9-Amino(9-deoxy)epi-cinchona alkaloid-tethered aluminium phosphonate architectures for heterogeneous cooperative catalysis: Asymmetric aldol and double-Michael cascade reaction. Mol. Cat. 2017, 434, 86–95. [Google Scholar] [CrossRef]
- Angeloni, M.; Piermatti, O.; Pizzo, F.; Vaccaro, L. Synthesis of Zirconium Phosphonate Supported L-Proline as an Effective Organocatalyst for Direct Asymmetric Aldol Addition. Eur. J. Org. Chem. 2014, 8, 1716–1726. [Google Scholar] [CrossRef]
- Wan, J.; Ding, L.; Wu, T.; Ma, X.; Tang, Q. Facile one-pot fabrication of magnetic nanoparticles (MNPs)-supported organocatalysts using phosphonate as an anchor point through direct co-precipitation method. RSC Adv. 2014, 4, 38323–38333. [Google Scholar] [CrossRef]
- Sabuzi, F.; Armuzza, V.; Conte, V.; Floris, B.; Venanzi, M.; Galloni, P.; Gatto, M. KuQuinones: A new class of quinoid compounds as photoactive species on ITO. J. Mater. Chem. C 2016, 4, 622–629. [Google Scholar] [CrossRef]
- Bonomo, M.; Sabuzi, F.; Di Carlo, A.; Conte, V.; Dini, D.; Galloni, P. KuQuinones as sensitizers of NiO based p-type dye sensitized solar cells. New J. Chem. 2017, 41, 2769–2779. [Google Scholar] [CrossRef]
- Volpato, G.A.; Marasi, M.; Gobbato, T.; Valentini, F.; Sabuzi, F.; Gagliardi, V.; Bonetto, A.; Marcomini, A.; Berardi, S.; Conte, V.; et al. Photoanodes for water oxidation with visible light based on a pentacyclic quinoid organic dye enabling proton-coupled electron transfer. Chem. Commun. 2020, 56, 2248–2251. [Google Scholar] [CrossRef] [PubMed]
- Valentini, F.; Sabuzi, F.; Conte, V.; Nemykin, V.N.; Galloni, P. Unveiling KuQuinone redox species: An electrochemical and computational cross study. J. Org. Chem. 2021, 86, 5680–5689. [Google Scholar] [CrossRef]
- Coletti, A.; Lentini, S.; Conte, V.; Floris, B.; Bortolini, O.; Sforza, F.; Grepioni, F.; Galloni, P. Unexpected One-Pot Synthesis of Highly Conjugated Pentacyclic Diquinoid Compounds. J. Org. Chem. 2012, 77, 6873–6879. [Google Scholar] [CrossRef] [Green Version]
- Sabuzi, F.; Lentini, S.; Sforza, F.; Pezzola, S.; Fratelli, S.; Bortolini, O.; Floris, B.; Conte, V.; Galloni, P. KuQuinones Equilibria Assessment for Biomedical Applications. J. Org. Chem. 2017, 82, 10129–10138. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, G.; Alauzun, J.G.; Granier, M.; Laurencin, D.; Mutin, P.H. Phosphonate Coupling Molecules for the Control of Surface/Interface Properties and the Synthesis of Nanomaterials. Dalton Trans. 2013, 42, 12569–12585. [Google Scholar] [CrossRef] [PubMed]
- Hagfeldt, A.; Boschloo, G.; Sun, L.; Kloo, L.; Pettersson, H. Dye-Sensitized Solar Cells. Chem. Rev. 2010, 110, 6595–6663. [Google Scholar] [CrossRef]
- Gardner, T.J.; Frisbie, C.D.; Wrighton, M.S. Systems for Orthogonal Self-Assembly of Electroactive Monolayers on Au and ITO: An Approach to Molecular Electronics. J. Am. Chem. Soc. 1995, 117, 6927–6933. [Google Scholar] [CrossRef]
- Lalatonne, Y.; Paris, C.; Serfaty, J.M.; Weinmann, P.; Lecouvey, M.; Motte, L. Bis-Phosphonates-Ultra Small Superparamagnetic Iron Oxide Nanoparticles: A Platform Towards Diagnosis and Therapy. Chem. Commun. 2008, 2553–2555. [Google Scholar] [CrossRef] [PubMed]
- Péchy, P.; Rotzinger, F.P.; Nazeeruddin, M.K.; Kohle, O.; Zakeeruddin, S.M.; Humphry-Baker, R.; Grätzel, M. Preparation of Phosphonated Polypyridyl Ligands to Anchor Transition-Metal Complexes on Oxide Surfaces: Application for the Conversion of Light to Electricity with Nanocrystalline TiO2 Films. J. Chem. Soc. Chem. Commun. 1995, 65–66. [Google Scholar] [CrossRef]
- Koh, S.E.; McDonald, K.D.; Holt, D.H.; Dulcey, C.S. Phenylphosphonic Acid Functionalization of Indium Tin Oxide: Surface Chemistry and Work functions. Langmuir 2006, 22, 6249–6255. [Google Scholar] [CrossRef]
- Michaelis, A.; Kaehne, R. Ueber das Verhalten der Jodalkyle gegen die sogen. Phosphorigsäureester oder O-Phosphine. Ber. Dtsch. Chem. Ges. 1898, 31, 1048–1055. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, A.K.; Thyagarajan, G. The Michaelis-Arbuzov Rearrangement. Chem. Rev. 1981, 81, 415. [Google Scholar] [CrossRef]
- Catel, Y.; Degrange, M.; Le Pluart, L.; Madec, P.J.; Pham, T.N.; Picton, L. Synthesis, Photopolymerization and Adhesive Properties of New Hydrolytically Stable Phosphonic Acids for Dental Applications. J. Polym. Sci. A Polym. Chem. 2008, 46, 7074–7090. [Google Scholar] [CrossRef]
- Ohashi, K.; Kosai, S.; Arizuka, M.; Watanabe, T.; Yamagiwa, Y.; Kamikawa, T. Syntheses of D-erythro-1-deoxydihydroceramide-1-sulfonic acid and phosphonosphingoglycolipid found in marine organisms via a common precursor. Tetrahedron 1989, 45, 2557–2570. [Google Scholar] [CrossRef]
- Ragulin, V.V. ω-Haloalkylphosphoryl Compounds: Synthesis and Properties. Russian, J. Gen. Chem. 2012, 82, 1928–1937. [Google Scholar] [CrossRef]
- Hewitt, D.G.; Newland, G.L. Organophosphorus compounds. P-arylated perhydro-1,2-azaphosphorines. Aust. J. Chem 1977, 30, 579–587. [Google Scholar] [CrossRef]
- Matoba, K.; Yonemoto, H.; Fukui, M.; Yamazaki, T. Structural modification of bioactive compounds. Syntheses of aminophosphonic acids. Chem. Pharm. Bull. 1984, 32, 3918–3925. [Google Scholar] [CrossRef] [Green Version]
- Groenewold, G.S.; Scott, J.R.; Lee, E.D.; Lammert, S.A. Rapid analysis of organophosphonate compounds recovered from vinyl floor tile using vacuum extraction coupled with a fast-duty cycle GC/MS. Anal. Methods 2013, 5, 2227–2236. [Google Scholar] [CrossRef]
- McLafferty, F.W. Mass spectrometric analysis. Molecular rearrangements. Anal. Chem. 1959, 31, 82–87. [Google Scholar] [CrossRef]
- Li, F.; Shishkin, E.; Mastro, M.A.; Hite, J.K.; Eddy, C.R.; Edgar, J.H.; Ito, T. Photopolymerization of Self-Assembled Monolayers of Diacetylenic Alkylphosphonic Acids on Group-III Nitride Substrates. Langmuir 2010, 26, 10725–10730. [Google Scholar] [CrossRef]
- Subedi, Y.P.; Alfindee, M.N.; Shrestha, J.P.; Becker, G.; Grilley, M.; Takemoto, J.Y.; Chang, C.W.T. Synthesis and biological activity investigation of azole and quinone hybridized phosphonates. Bioorg. Med. Chem. Lett. 2018, 28, 3034–3037. [Google Scholar] [CrossRef]
- Villemin, D.; Simeon, F.; Decreus, H.; Jaffres, P.A. Rapid and efficient arbuzov reaction under microwave irradiation. Phosphorus Sulfur Silicon Relat. Elem. 1998, 133, 209–213. [Google Scholar] [CrossRef]
- Cherevatskaya, M.; Neumann, M.; Füldner, S.; Harlander, C.; Kümmel, S.; Dankesreiter, S.; Pfitzner, A.; Zeitler, K.; König, B. Visible-Light-Promoted Stereoselective Alkylation by Combining Heterogeneous Photocatalysis with Organocatalysis. Angew. Chem. Int. Ed. 2012, 51, 4062–4066. [Google Scholar] [CrossRef]
- Sevrain, C.M.; Berchel, M.; Couthon, H.; Jaffrès, P.A. Phosphonic acid: Preparation and applications. Beilstein J. Org. Chem. 2017, 13, 2186–2213. [Google Scholar] [CrossRef] [PubMed]
- Ikenberry, M.; Peña, L.; Wei, D.; Wang, H.; Bossmann, S.H.; Wilke, T.; Wang, D.; Komreddy, V.R.; Rillema, D.P.; Hohn, K.L. Acid monolayer functionalized iron oxide nanoparticles as catalysts for carbohydrate hydrolysis. Green Chem. 2014, 16, 836–843. [Google Scholar] [CrossRef] [Green Version]
- Jansa, P.; Baszczyňski, O.; Procházková, E.; Dračìnský, M.; Janeba, Z. Microwave-assisted hydrolysis of phosphonate diesters: An efficient protocol for the preparation of phosphonic acids. Green Chem. 2012, 14, 2282–2288. [Google Scholar] [CrossRef]
- Besse, V.; Le Pluart, L.; Cook, W.D.; Pham, T.N.; Madec, P.J. Synthesis and polymerization kinetics of acrylamide phosphonic acids and esters as new dentine adhesives. J. Polym. Sci. A Polym. Chem. 2013, 51, 149–157. [Google Scholar] [CrossRef]
- Norris, M.R.; Concepcion, J.J.; Glasson, C.R.K.; Fang, Z.; Lapides, A.M.; Ashford, D.L.; Templeton, J.L.; Meyer, T.J. Synthesis of Phosphonic Acid Derivatized Bipyridine Ligands and Their Ruthenium Complexes. Inorg. Chem. 2013, 52, 12492–12501. [Google Scholar] [CrossRef]
- Zhou, Y.; Ayad, S.; Ruchlin, C.; Posey, V.; Hill, S.P.; Wu, Q.; Hanson, K. Examining the Role of Acceptor Molecule Structure in Self-Assembled Bilayers: Surface Loading, Stability, Energy Transfer, and Upconverted Emission. Phys. Chem. Chem. Phys. 2018, 20, 20513–20524. [Google Scholar] [CrossRef]
- Morita, T.; Okamoto, Y.; Sakurai, H. A convenient dealkylation of dialkyl phosphonates by chlorotrimethylsilane in the presence of sodium iodide. Tetrahedron Lett. 1978, 28, 2523–2526. [Google Scholar] [CrossRef]
- Franz, R.G. Comparisons of pKa and log P values of some carboxylic and phosphonic acids: Synthesis and measurement. AAPS Pharm. Sci. 2001, 3, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, K.D.; Andreopoulou, A.K.; Kallitsis, J.K. Phosphonated fully aromatic polyethers for PEMFCs applications. J. Polym. Sci. A Polym. Chem. 2010, 48, 2817–2827. [Google Scholar] [CrossRef]
- Kahraman, G.; Wang, D.Y.; Von Irmer, J.; Gallei, M.; Hey-Hawkins, E.; Eren, T. Synthesis and Characterization of Phosphorus- and Carborane-Containing Polyoxanorbornene Block Copolymers. Polymers 2019, 11, 613–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Scheme 1.
Retrosynthetic analysis to obtain new KuQuinone phosphonic acid derivatives [32].
Scheme 1.
Retrosynthetic analysis to obtain new KuQuinone phosphonic acid derivatives [32].

Scheme 2.
Diethyl ω-bromoalkylphosphonates synthesis.

Scheme 3.
(a): Arbuzov reaction for the synthesis of 3; (b–d) concurrent processes: diethyl ethylphosphonate generation (b), di-phosphonation (c) and cyclization (d).
Scheme 3.
(a): Arbuzov reaction for the synthesis of 3; (b–d) concurrent processes: diethyl ethylphosphonate generation (b), di-phosphonation (c) and cyclization (d).

Scheme 4.
Synthesis of KuQuinone phosphonate esters 4–6.

Scheme 5.
Hydrolysis of KuQuinone phosphonate esters.

Figure 1.
31P NMR spectra of 8 (purple) and 5 (green) in DMSO-d6.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Optimization of reaction conditions for the synthesis of diethyl ω-bromoalkylphosphonate. Conditions: α,ω-dibromoalkane (75 mmol): triethyl phosphite (75 mmol); T = 140 °C.
Table 1.
Optimization of reaction conditions for the synthesis of diethyl ω-bromoalkylphosphonate. Conditions: α,ω-dibromoalkane (75 mmol): triethyl phosphite (75 mmol); T = 140 °C.
| Entry | Br(CH2)nBr | Reaction Time | Isolated Yield (%) |
|---|---|---|---|
| 1 | n = 4 | 12 h | - |
| 2 a | n = 4 | 12 h | - |
| 3 b | n = 4 | 5 min | - |
| 4 a,c | n = 6 | 90 min | 5 |
| 5 a,d | n = 6 | 3 h | 40 |
| 6 a,d | n = 5 | 3 h | 40 |
| 7 a,d | n = 4 | 3 h | 20 e |
a Bromoethane distilled during the reaction. b microwave irradiation at 150 W. c triethyl phosphite dropwise addition within 30 min. d triethyl phosphite dropwise addition within 2 h. e isolated by column chromatography (ethyl acetate/Al2O3).
Table 2.
Synthesis of KuQuinone diethyl phosphonate derivatives. Reaction conditions: 5.75 mmol of 2-hydroxy-1,4-naphthoquinone, 12 mmol of bromoalkylphosphonate, 8 mmol of Cs2CO3, 0.33 mmol of FcH, in 22 mL of DMSO, at 114 °C, for 41 h.
Table 2.
Synthesis of KuQuinone diethyl phosphonate derivatives. Reaction conditions: 5.75 mmol of 2-hydroxy-1,4-naphthoquinone, 12 mmol of bromoalkylphosphonate, 8 mmol of Cs2CO3, 0.33 mmol of FcH, in 22 mL of DMSO, at 114 °C, for 41 h.
| Reagent | Product | Yield (%) |
|---|---|---|
| 1 | 4 | 11 |
| 2 | 5 | 10 |
| 3 | 6 | 7 |
Table 3.
Hydrolysis of KuQuinone phosphonate esters. Conditions: 0.1 mmol of diethyl phosphonate KuQuinone, 5 mmol of NaI, 5 mmol of bromotrimethylsilane, in 16 mL of dry CH3CN: CHCl3 (1: 1), at 40 °C, 4 h, under N2 atmosphere. Then, silyl ester hydrolysis was performed with 30 mL of methanol.
Table 3.
Hydrolysis of KuQuinone phosphonate esters. Conditions: 0.1 mmol of diethyl phosphonate KuQuinone, 5 mmol of NaI, 5 mmol of bromotrimethylsilane, in 16 mL of dry CH3CN: CHCl3 (1: 1), at 40 °C, 4 h, under N2 atmosphere. Then, silyl ester hydrolysis was performed with 30 mL of methanol.
| Reagent | Product | Yield (%) |
|---|---|---|
| 4 | 7 | 100 |
| 5 | 8 | 95 |
| 6 | 9 | 97 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Forchetta, M.; Conte, V.; Fiorani, G.; Galloni, P.; Sabuzi, F. A Sustainable Improvement of ω-Bromoalkylphosphonates Synthesis to Access Novel KuQuinones. Organics 2021, 2, 107-117. https://doi.org/10.3390/org2020010
AMA Style
Forchetta M, Conte V, Fiorani G, Galloni P, Sabuzi F. A Sustainable Improvement of ω-Bromoalkylphosphonates Synthesis to Access Novel KuQuinones. Organics. 2021; 2(2):107-117. https://doi.org/10.3390/org2020010
Chicago/Turabian StyleForchetta, Mattia, Valeria Conte, Giulia Fiorani, Pierluca Galloni, and Federica Sabuzi. 2021. "A Sustainable Improvement of ω-Bromoalkylphosphonates Synthesis to Access Novel KuQuinones" Organics 2, no. 2: 107-117. https://doi.org/10.3390/org2020010