Borate Minerals and RNA Stability


1
Dipartimento di Biologia e Biotecnologie “Charles Darwin”, Università di Roma, “Sapienza” Rome, Italy
2
Dipartimento di Scienze e Tecnologie Chimiche, Università “Tor Vergata” 00133 Rome, Italy
3
Dipartimento ABAC, Università della Tuscia, Viterbo, Italy
*
Author to whom correspondence should be addressed.
Polymers 2010, 2(3), 211-228; https://doi.org/10.3390/polym2030211
Submission received: 21 May 2010
/
Revised: 7 July 2010
/
Accepted: 12 August 2010
/
Published: 16 August 2010
(This article belongs to the Special Issue Natural Polymers)
Abstract
:The abiotic origin of genetic polymers faces two major problems: a prebiotically plausible polymerization mechanism and the maintenance of their polymerized state outside a cellular environment. The stabilizing action of borate on ribose having been reported, we have explored the possibility that borate minerals stabilize RNA. We observe that borate itself does not stabilize RNA. The analysis of a large panel of minerals tested in various physical-chemical conditions shows that in general no protection on RNA backbone is exerted, with the interesting exception of ludwigite (Mg2Fe3+BO5). Stability is a fundamental property of nucleic polymers and borate is an abundant component of the planet, hence the prebiotic interest of this analysis.
1. Introduction
The intense debate on the nature of life has led to the minimalist definition: “a self-sustaining chemical system capable to undergo replication and Darwinian evolution” [1]. Each of the four concepts onto which this definition relies (self-sustenance, chemical information, replication, evolution) entails the existence of an informational polymer endowed of genetic functions. This function, now plied on Earth in the vast majority of living systems by DNA, presumably originated as RNA. The “RNA world” theory [2,3,4] is based on the double nature of RNA, carrier of both genetic information and of the catalytic functions [5,6] from which RNA-mediated synthesis of other RNAs evolved. Even though macromolecular nucleic information may be organized in a vast number of alternative structures [7,8,9,10] extant nucleic acids have no natural substitutes. This very fact points to the relevance of the problems related to the origin and to the stability of their constituents.
Focusing on the sugar moiety of extant nucleic acids, one mayor objection to the plausibility of the RNA world theory is the instability of ribose [4]. This property would have presumably prevented its accumulation under prebiotic conditions, or at least decreased its presence diminishing the centrality of its initial function. A possible solution is provided by the observation of the stabilization of adenosine by borate [11] and of ribose by borate and by borate minerals ulexite, kernite and colemanite [12]. The borate complex of ribose is more stable than the complexes of related aldopentoses [11,12,13]. A theoretical study of the factors controlling the stability of the borate complexes of ribose, arabinose, lyxose and xylose [14] confirmed these observations and revealed that in the aldopentoses-borate complexes, the electrostatic field of the borate is strong enough to change the orientation of the hydroxyl groups, causing increased stability. A coherent structural model of the ribose-borate 2:1 stable complex was defined [14].
Is the boron protective effect observed for ribose [11,12,13] and for a nucleoside [11] instrumental also for the protection of nucleosides when present in the ribonucleic polymerized form? This question remains unexplored. Thus, we have analyzed the effect of boric acid, of tetraborate and of a large collection of borate minerals on the stability of RNA oligomers.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mineral | Formula | MW |
|---|---|---|
| Axinite-(Mn) | Ca2Mn2+Al2(BO3)Si4O12(OH) | 569.21 |
| Canavesite | Mg2(CO3)(HBO3)•5(H2O) | 258.51 |
| Chambersite | Mn2+3B7O13Cl | 483.93 |
| Colemanite | Ca2B6O11•5(H2O) | 411.09 |
| Dravite | NaMg3Al6(BO3)3Si6O18(OH)4 | 958.75 |
| Dumortierite | Al6.9(BO3)(SiO4)3O2.5(OH)0.5 | 569.73 |
| Elbaite | NaLi2.5Al6.5(BO3)3Si6O18(OH)4 | 916.68 |
| Hambergite | Be2(BO3)(OH) | 93.84 |
| Hydroboracite | CaMgB6O8(OH)6•3(H2O) | 413.33 |
| Jeremejevite | Al6B5O15F2.5(OH)0.5 | 511.93 |
| Johachidolite | CaAl(B3O7) | 211.49 |
| Kernite | Na2B4O6(OH)2·3(H2O) | 290.28 |
| Kornerupine | (Mg,Fe2+)4(Al,Fe3+)6(SiO4,BO4)5(O,OH)2 | 734.04 |
| Kurnakovite | MgB3O3(OH)5•5(H2O) | 279,85 |
| Ludwigite | Mg2Fe3+BO5 | 195.26 |
| Painite | CaZrB[Al9O18] | 586.42 |
| Rhodizite | (K,Cs)Al4Be4(B,Be)12O28 | 778.83 |
| Schorl | NaFe2+3Al6(BO3)3Si6O18(OH)4 | 1,053.38 |
| Ulexite | NaCaB5O6(OH)6•5(H2O) | 405.23 |
| Vonsenite | Fe2+2Fe3+BO5 | 258.35 |
2. Results and Discussion
2.1. The RNA stability assay
The effect of borate and borate minerals was analyzed both in water and in formamide at 80 °C. This particular physical-chemical setting was selected because RNA degradation in these conditions takes reasonable experimental time span, as previously detailed [15,16], and because of its per se interest in a prebiotic perspective [17]. A preliminary set of analyses consisted in (i) the determination of the stability of borate minerals at 80 °C in water or in formamide and in (ii) the measurement of the variation of pH induced by minerals in the aqueous environment. The borate minerals studied are listed in Table 1. The results of this analysis are reported in Table 2. Stability of the minerals was determined by measuring with the curcumin colorimetric assay the boron released in the conditions of the RNA stability assay: 80 °C in water or in formamide.
| Mineral | Boron released1 | pH2 | |||||
|---|---|---|---|---|---|---|---|
| H2O | H2NCOH | ||||||
| 4 hrs | 18 hrs | 4 hrs | 18 hrs | 24 °C | 80 °C | ||
| Ca2Mn2+Al2(BO3)Si4O12(OH) | 1.37 | 2.90 | 0 | 0 | 5.82 | 5.45 | |
| 3.68 | 10.38 | 6.07 | 7.03 | 8.00 | 8.31 | ||
| 0.26 | 1.26 | 0.07 | 0.19 | 6.62 | 6.27 | ||
| 12.52 | 43.87 | 9.07 | 17.75 | 7.38 | 7.56 | ||
| 0.77 | 1.80 | 0 | 0 | 6.12 | 5.72 | ||
| 0.19 | 0.46 | 0 | 0 | 7.15 | 7.20 | ||
| 0.81 | 1.59 | 0 | 0 | 6.18 | 6.26 | ||
| 0.07 | 0.21 | 0 | 0 | 6.68 | 6.02 | ||
| 0.03 | 0.74 | 0 | 0 | 8.26 | 8.61 | ||
| 0.15 | 0.38 | 0 | 0 | 6.38 | 5.82 | ||
| 1.01 | 1.76 | 0 | 0 | 6.21 | 6.14 | ||
| 2.85 | 3.73 | 100.00 | 100.00 | 8.47 | 8.36 | ||
| 0.36 | 0.69 | 0 | 0 | 5.72 | 5.39 | ||
| 2.16 | 5.84 | 0 | 0 | 7.53 | 7.68 | ||
| 0.07 | 0.30 | 0 | 0 | 7.02 | 6.85 | ||
| 1.34 | 2.88 | 0 | 0 | 5.81 | 5.31 | ||
| 0.03 | 0.03 | 0 | 0 | 6.78 | 6.50 | ||
| 0.092 | 0.63 | 0 | 0 | 5.79 | 5.38 | ||
| 25.07 | 70.59 | 3.69 | 8.61 | 7.38 | 7.72 | ||
| 0.73 | 0.81 | -- | 0 | 6.26 | 5.87 | ||
| 5.313 | 4.883 | ||||||
| 5.333 | 4.743 | ||||||
1 Boron released at 80 °C after the indicated time. The values are reported as % of the amount of boron initially present in the mineral; 2 pH measurement after 4 hours at the indicated temperature; 3 Measured at 100 mM; 4 The minerals colemanite, kernite, kurnakovite and ulexite were found to be rapidly solubilized, thus releasing larger amounts of boron. For these minerals the assay was scaled down 10fold, starting with 0.1 mg/mL instead of 1.0 mg/mL. Mean standard deviation: Boron release = ±0.03 for the % values up to 5.84, 0.5% for the % values between 15 and 100%. pH = ±0.06.
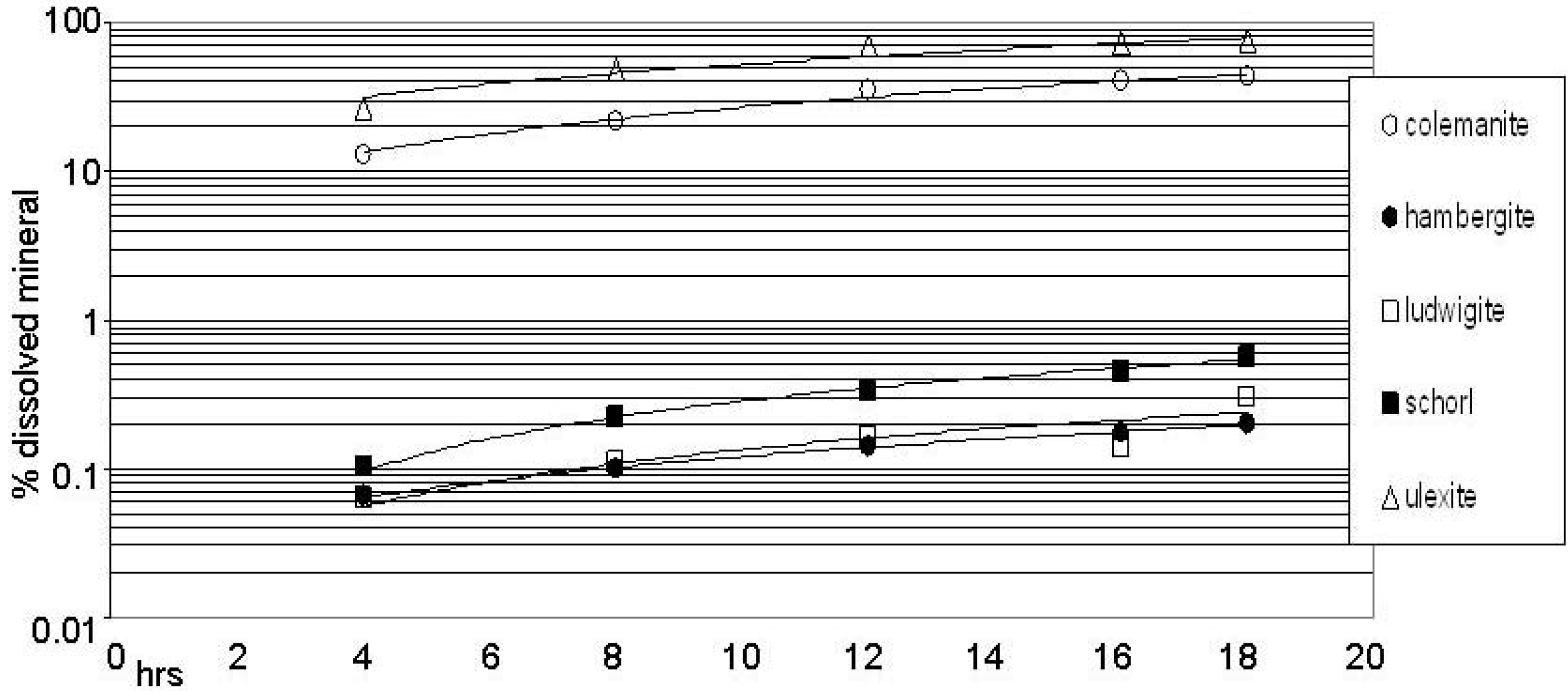
Figure 1 shows selected instances of boron release from minerals (%, 80 °C, water, note the logarithmic vertical scale) relative to the total amount initially present in mineral form. The minerals shown are representative of fast (colemanite, ulexite) and slow (hambergite, schorl, ludwigite) dissolution.
Figure 1.
Boron release in water.

The amount is shown of boron released upon treatment in water of the indicated minerals, as determined by the curcumin colorimetric assay. The assay was performed on 1 mg/mL for hambergite, ludwigite and schorl, on 0.1 mg/mL for colemanite and ulexite (see also legend to Table 2). The lines connecting the experimental points, here and in the following plots, have no mathematical meaning. They are given only to facilitate identification. Mean standard deviation: see legend to Table 2.
Table 2 reports the % of boron released at two representative time points (4 and 18 hrs) in water for all the minerals analyzed. Data for the dissolution in formamide are also reported. In conclusion, in water boron was released, although with quite different kinetics, by all the borates analyzed. Only rhodizite remained undissolved, within the error limit of the assay. Faster dissolutions were observed for colemanite, kernite, kurnakovite and ulexite. In formamide the majority of minerals showed high stability with the exception of canavesite, colemanite and ulexite. Noteworthy, kernite dissolved immediately and completely. The pH variation as measured in the RNA stability assay conditions is reported in the two rightmost columns of Table 2. No extreme variations were observed.
The stability of RNA in the presence of boron or borate minerals in water or in the formamide organic environment was measured by treatment with these compounds of homogeneous (PolyA24) or heterogeneous (P1) 5’-terminally labelled ribooligonucleotide. The RNA remaining after treatment was analyzed by gel electrophoresis. The eventual stable interaction of RNA with mineral surfaces was also determined.
2.2. RNA stability in boron minerals
The effect of borates on RNA stability was studied as follows. First the stability of PolyA24 and P1 ribonucleotides in water and in formamide at 80 °C was determined. Figure 2 Panel A shows the kinetics of hydrolytic degradation of PolyA24 in water at 80 °C. The pattern of degradation shows that the 24mer was regularly hit by low-occurrence hydrolytic events along its whole length, as shown by the appearance of ladder-like degradation profiles. After about 10 hours in water, faster degradation of the RNA started. The relatively high stability of the PolyA sequence in aqueous solution is well established [18,19,20]. The stability of RNA phosphodiester bonds has repeatedly been associated with the stacking interactions between adjacent bases [21,22,23]. Nucleic acid base-stacking is at present an understood phenomenon [24,25,26,27,28]. The possibility that the initial stability of the PolyA oligonucleotide towards hydrolysis is a base stacking-related effect was discussed in [15].
Figure 2.
Instability of RNA oligonucleotides in water and in formamide.

Panel A: Degradation kinetics of PolyA24 as a function of time (hours, minutes: h.mm) at 80 °C in water and (Panel B) in formamide. 5’ Labelled oligonucleotide was treated (see Experimental Section) for the period of time indicated on top of each lane and analyzed in 16% acrylamide gel electrophoresis. Numbering of fragment sizes is on both left and right sides. U = untreated. As reported [15], the subterminal phosphodiester bond on the 3’ extremity of the molecule (the 23d) is more resistant to hydrolysis, causing the absence of the 23mer from the degradation ladder-wise profile. The bands representing the 2’-3’ cyclic phosphate and the 2’ or 3’ free phosphate extremities are indicated (right of panel A, bottom). For this attribution, see (15, and references therein). In water degradation started after a lag period of about 10 hours (as described and discussed in [15]), while in formamide the degradation kinetics follows a first order kinetics since the beginning.
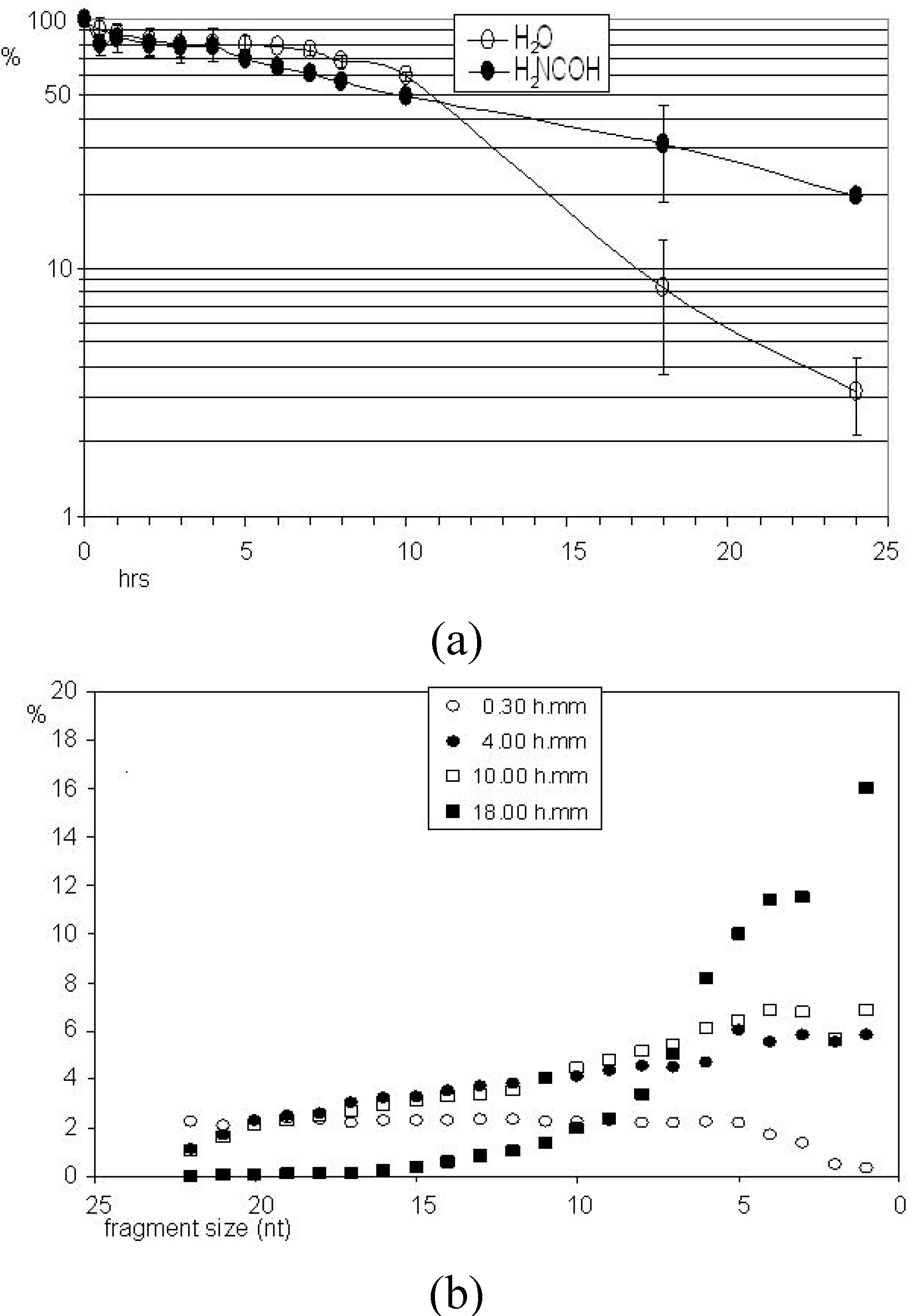
Figure 2 Panel B shows the kinetics of degradation of PolyA24 in formamide. The degradation profile proceeds regularly, with a typical linear progression, favored by the formamide-induced unstacking of nucleic bases. The mechanism of degradation of RNA by formamide is known [29]. Figure 3 shows a quantitative description of these degradations. Panel A reports the percentage (%) of intact molecules (ordinate) as a function of time (abscissa) in both water and formamide. Panel B describes the progression of the hydrolytic process in water at selected time points showing a regular progression. Hence the absence of non-linearities caused by fragment-length, terminal, sub-terminal or sequence context-induced effects. The same behaviour was observed for the mixed-sequence P1 RNA (not shown).
Figure 3.
Quantitative analysis of the degradation of the full-length molecules reported in Figure 2.
Figure 3.
Quantitative analysis of the degradation of the full-length molecules reported in Figure 2.

Panel (a): the molecules remaining intact are given as a percentage (ordinate) of the full-sized molecules (uppermost band) present at each time point (abscissa) relative to the zero time control. Average of two experiments, with error bars. Panel (b) quantitative evaluation of the progression of hydrolysis. The percentage is reported of each hydrolytically-produced band relative to the total (100%) material present in the sample lane. Representative time points are reported (30 min, 4, 10 and 18 hours), as indicated. Allowance made for an average error of 1% (not indicated for graphical reasons) the plot shows the regularity of the degradation process.
A general first screening of the effect of borates on RNA was performed as follows: 2 pmoles of a 5’ end-labelled oligonucleotide (PolyA24 or P1) were reacted in water with three different concentrations of boric acid, of sodium tetraborate, or of one of the 20 minerals considered (see list in Experimental Section), namely with 10, 102 or 103 µg/mL. The reaction was allowed for different time periods, usually 3 × 10, 1.8 × 102 and 1.08 × 103 min. For the minerals that caused modification of degradation kinetics (both faster degradation or enhanced half-life of RNA) relative to that observed in the controls in their absence, the kinetics were analyzed in greater detail.
Figure 4 details the stability of P1 RNA in boric acid. Panel A shows the kinetics of hydrolysis (0, 30 min, 3 and 18 hours, as indicated) in water and in 100 mM boric acid, tested at the indicated pH values. Boric acid (whose addition to water induces at 80 °C the pH value 5.0) does not protect RNA (cfr lanes 1–4 with lanes 5–8). Panel B shows the corresponding plot.
Figure 4.
RNA stability in boric acid.

P1 RNA was reacted in water and boric acid in various conditions, analyzed by gel electrophoresis and quantitatively evaluated. Panel A: treatment for the indicated times (0, 30 min, 3 and 18 hours, as indicated) at 80 °C at the indicated pH values in the absence (lanes 1–4) or in the presence of 100 mM boric acid (lanes 5–16). Panel B: % of the full-length molecules remaining intact after the indicated time in H2O or boric acid, as indicated. Data from Panel A. Panel C: full-sized molecules after treatment in water at the indicated pH values obtained with Tris-HCl. Time points as in panel A. Panel D: same in boric acid, at the indicated concentrations, pH 5.0.
Panel C shows the degradation kinetics (same time points, same pH range of values as those reported in panel A) in water, without boric acid. The comparison of the bands corresponding to full-sized molecules (indicating the first-hit kinetics of macromolecular degradation) of Panel A with panel C shows that boric acid does not protect RNA at none of the pH values tested. Panel D shows that in a range of two orders of magnitude of boric acid concentration (keeping the pH fixed at 5.0) no protection is observed.
2.3. The effect of minerals on RNA stability
Figure 5 shows five examples of the “4 × 4” assay (four time points per four mineral concentrations) run on PolyA24 in water or in formamide. The examples shown were selected as representative of: Panel A, increased degradation in water (canavesite). Panel B, protection by low concentration of mineral in water (jeremejevite). Panel C, increased degradation in formamide (hydroboracite). Panel D, protection in formamide (rhodizite). Similar results were obtained P1 RNA in formamide, showing the sequence-independence of the protective and of the degradative effects. All the minerals were tested with this assay. The results, summarized in Table 3, showed that the large majority of the minerals analyzed caused faster degradation of the polymeric form, both in water and in formamide. Figure 6 shows a more detailed kinetic analysis on ludwigite. The same was performed (at the three concentrations selected) on all the minerals that, according to the data reported in Table 3, hinted to a protective effect.
Figure 5.
RNA stability in water and in formamide in the presence of borate minerals. The assay was run as described in Figure 2 in the presence of the indicated mineral: canavesite (panel A), jeremejevite (panel B), hydroboracite (panel C), rhodizite (panel D) in water (panels A–B) and formamide (panel C–D) for the indicated time spans (0, 0.30, 3 and 18 hours) with the indicated mineral.
Figure 5.
RNA stability in water and in formamide in the presence of borate minerals. The assay was run as described in Figure 2 in the presence of the indicated mineral: canavesite (panel A), jeremejevite (panel B), hydroboracite (panel C), rhodizite (panel D) in water (panels A–B) and formamide (panel C–D) for the indicated time spans (0, 0.30, 3 and 18 hours) with the indicated mineral.

| H2O | Formamide | H2O | Formamide | H2O | Formamide |
|---|---|---|---|---|---|
| No effect | Protection | Degradation | |||
| Axinite-(Mn) | Jeremejevite * | Rhodizite 1.8 × 103 | Kurnakovite 6 | Canavesite 6.5 × 10 | |
| Colemanite | Ludwigite * | Elbaite 1.5 × 103 | Hydroboracite 8 | Kurnakovite 1.05 × 102 | |
| Dravite | Hambergite * | Ulexite 10 | Hambergite 1.15 × 102 | ||
| Dumortierite | Dumortierite 3 × 10 | Ulexite 1.9 × 102 | |||
| Jeremejevite | Chambersite 3.6 × 10 | Kernite 2 × 102 | |||
| Johachidolite | Axinite-(Mn) 6.5 × 10 | Hydroboracite 2.8 × 102 | |||
| Kornerupine | Kornerupine 7.5 × 10 | Chambersite 3.6 × 102 | |||
| Painite | Schorl 7.8 × 10 | Ludwigite 4 × 102 | |||
| Schorl | Kernite 8 × 10 | ||||
| Vorsenite | Elbaite 1.05 × 102 | ||||
| Vonsenite 1.1 × 102 | |||||
| Painite 1.2 × 102 | |||||
| Canavesite 1.4 × 102 | |||||
| Johachidolite 2.2 × 102 | |||||
| Rhodizite 2.4 × 102 | |||||
| Colemanite 2.5 × 102 | |||||
Minerals are listed in order of decreasing effect, both for protection and for degradation. The number after each mineral name gives the t½ of the 3’ phosphoester bond (minutes) at 1 mg/mL of mineral in the assay. Standard deviation: ±3% of the t½ value; * jeremejevite, ludwigite and hambergite display a double effect: protection at low dose of mineral and enhanced degradation at high dose. The observed t½ values are: (1) 3.3 × 103 min at 0.01 mg/mL, 1.7 × 102 min at 1 mg/mL; (2) 1.16 × 103 at 0.01 mg/mL and 102 at 1 mg/mL; (3) 1.10 × 103 at 0.01 mg/mL and 3.6 × 10 at 1 mg/mL, respectively.
Figure 6.
RNA protection in water by ludwigite, performed as described in Figure 2 and Figure 5, for the indicated time spans (panel A). The plot in panel B shows the remaining full-length molecules (%) at the indicated time in the presence (•) or in the absence (○) of ludwigite.

The three minerals whose interaction with RNA in water increased stability (jeremejevite, ludwigite, hambergite) display a common behaviour: stabilization is observed only at low mineral concentration (10 µg/mL) while at high mineral concentration (103 µg/mL) degradation is stimulated. These three minerals do not have common elemental composition (in addition to boron, jeremejevite contain Al and F; ludwigite Mg and Fe; hambergite Be) suggesting that protection could be provided (i) by the only common element, boron, upon its possible release or (ii) by interaction of the RNA oligomer with mineral surfaces. Given that free boron does not directly protect RNA (Figure 4) this second possibility is favourably considered (see Discussion).
2.4. The retention of RNA by minerals
Both PolyA24 and P1 ribooligonucleotides were treated with the crushed minerals and the RNA absorbed was measured relative to that remaining in solution. Data were obtained as described in Experimental Section.
The assay, performed on the 12/20 minerals that do not rapidly dissolve at 80 °C (as reported in Table 3), showed that all the minerals adsorb RNA, the more efficient (>80% most absorption at 4 hours) being aixinite-(Mn), elbaite, hambergite, hydroboracite, ludwigite, schorl. After 18 hours a large part (>70%) of the initially adsorbed material remained bound only to elbaite, hambergite and hydroboracite. No difference was observed between PolyA24 and P1 RNA. Given its protective capacity, jeremejevite was also analyzed.
3. Experimental Section
3.1. Minerals
Given the high elemental complexity of several minerals (i.e., schorl, kornerupine, etc.) preventing any factual ordering, the list is organized alphabetically. The minerals analysed here were selected on the basis of their representative elemental composition, their availability, and the lack of appreciable endogenous radioactivity. Only rhodizite is barely detectably radioactive (Gamma Ray Petroleum Institute Units = 52.60). The minerals were obtained from Ezio Curti ([email protected]), former provider and consultant of the Collection of Minerals of the Department of Mineralogy (University of Rome “Sapienza”). Homogenous material was isolated under the microscope, washed twice (first with ethanol, then with analytical grade distilled water), air dried, then manually ground in a ceramic mortar. Samples of crushed materials are available upon request, except for canavesite, chambersite and kornerupine, which may be purchased from Greenside Minerals (http://www.greensideminerals.com), Dakota Matrix Minerals (http://www.dakotamatrix.com) or John Betts Fine Minerals (www.johnbetts-fineminerals.com). Boric acid (H3BO3, SigmaUltra ≥99.5%), sodium tetraborate (Na2B4O7) and curcumin were from Sigma Aldrich. H2O was bidistilled deionized by a MilliQ apparatus (MΩ.cm at 25 °C), then sterilized. Formamide was from Fluka (>99%).
3.2. The RNA oligomers used
The degradation of ribonucleotides by water or formamide was studied by using the 24mer 5’-AAAAAAAAAAAAAAAAAAAAAAAA-3’ (PolyA24) or the 20mer 5′-GGAAACGUAUCCUUUGG GAG-3′ dubbed P1 [41], purchased from Dharmacon Inc. (Chicago, IL, USA). PolyA24 was selected because of its homogeneity, while P1 was studied for the opposite reason. Its sequence encompasses 14 of the 16 possible base-steps, with the exclusion of the two unstable GpC and CpA steps. The comparison of the degradation profiles obtained for the homogeneous versus the heterogeneous sequence potentially provides information on sequence-specificity effects induced by boron.
3.3. RNA preparation and 5’ labelling
PolyA24 and P1 RNA were labeled with [γ-32P]ATP using polynucleotide kinase (Roche Applied Science). The oligonucleotides were then purified on a 16% denaturing acrylamide gel (19:1 acrylamide/bisacrylamide). After elution, the residual polyacrylamide was removed by a NuncTrap Probe purification column (Stratagene). Two pmol (typically 30,000 cpm) RNA were processed for each sample.
3.4. The RNA stability assay
The degradation of RNA oligonucleotides in water is a well studied process [15,30,31,32]. The degradation reaction in formamide was also described [16,17 and refs. therein]. The cleavage of the phosphoester chain in water normally requires participation of the 2’-OH group as an internal nucleophile [33] in two “nucleophilic cleavage” events: the transesterification and hydrolysis reactions. During transesterification, the 2’-OH nucleophile attacks the tetrahedral phosphorus to afford a 2’,3’ cyclic monophosphate. This species is then hydrolyzed into a mixture of 2’- and 3’-phosphate monoesters. Both steps are, more in general, catalysed by protons, hydroxide, nitrogen derivatives and metal ions. The two molecular species (2’,3’-cyclic phosphate and 2’- or 3’-phosphate) can be separated in analytical electrophoretic gels (i.e., see the lower part of Figures 2A and B). The information provided by this assay pertains to: (i) the stability of the 3’ phosphoester bond, the weakest bond in RNA both in water and in formamide [16]; (ii) the rate of 2’,3’ cyclic phosphate bridge opening.
The 5′-labeled oligonucleotide was treated under the time, temperature, and solution conditions indicated where appropriate. A typical assay consisted of 75 μL containing 5 μL of RNA solution, 5–20 μL of mineral (solution or suspension of ground material) in water or formamide (H2NCOH) as appropriate, complement of water or formamide to 75 μL. To stop the reaction a solution of 5 × 10−4 M (final concentration) of tetrasodium pyrophosphate (Sigma) dissolved in water, pH 7.5, was added to a final volume of 40 μL. The samples were vortexed for 1 min, then centrifuged at 13,000 rpm for 20 min (Haereus Biofuge). This procedure was performed twice. The supernatant was ethanol precipitated, resuspended in 5 μL of formamide buffer, heated for 2 min at 65 °C, and loaded on a 16% denaturing polyacrylamide gel (19:1 acrylamide/bisacrylamide). For further details, see [16].
The half-life of the oligonucleotide was determined with standard graphical procedure from plots of the % disappearance of the intact 20- or 24-mer molecules. Given that one disappearing molecule represents one cleavage, and given the verification of the two basic assumptions (no 5′-cleavage, no sequence-biased preferential cleavage), the half-life of the single 3′-phosphoester bonds in the ribooligonucleotide is given by the half-life of the oligonucleotide ×23 or ×19 (that is: the number of 3′-5’ phosphodiester bonds in the 24-mer or in 20-mer, respectively).
3.5. Measurement of released boron
Released boron was measured by the curcumin colorimetric assay [34], as follows: 10 mg of powdered mineral were suspended in 1 mL of H2O and treated for the indicated time (usually between 0 and 18 hrs) at the desired temperature. Samples were centrifuged (15 min, 13,000 rpm), the supernatant was dried in oven, resuspended in 2 mL of curcumin solution, dried at 55 °C under aspiration. To each dried sample 5 mL of 95% ethanol were added, followed by stirring and resulting in the solubilization of the red/orange precipitate. The sample was diluted 1:25 with 95% ethanol and the OD at 540 mm was determined against blanks processed in parallel. The curcumin solution (0, 40 g/L) consisted of 80 mg of curcumin in 10.0 g of dehydrated oxalic acid added to 150 mL ethanol. 8,4 mL chloride acid were added, followed by dilution to 200 mL with 95% ethanol. The solution is stable for a week a 4 °C. Titration plots performed on boric acid (H3BO3) allowed the appropriate conversion from OD540 to the actual amount of released boron. Taking into account the relative quantitative presence of boron in the mineral under consideration according to the composition formulas reported above, the boron released by mineral dissolution was determined.
3.6. Measurement of RNA retained by minerals
2 pmoles (30,000 cpm) of 5’ terminally labelled RNA were treated with ground mineral (100 µg) in 75 µL of water at 80 °C for 4 hrs with periodical stirring followed by centrifugation (13,000 rpm, 30 min). The amount of material remaining associated with the precipitate and that in solution were measured (value: 4 hrs). The RNA-precipitated mineral was re-dissolved and treated similarly for additional 14 hrs. The measurements were then repeated similarly (value: 18 hrs) (see Table 4).
Table 4.
RNA retention % on borate minerals. 2 pmoles (30,000 cpm) of P1 RNA were treated with the indicated mineral, as described in Methods. The amount of RNA retained by the mineral at 4 and 18 hrs is reported as percentage of the RNA input.
| Mineral | 4 hrs | 18 hrs |
|---|---|---|
| Axinite-(Mn) | 84 | 56 |
| Dravite | 72 | 44 |
| Dumortierite | 69 | 41 |
| Elbaite | 93 | 79 |
| Hambergite | 89 | 72 |
| Hydroboracite | 90 | 76 |
| Kornerupine | 76 | 44 |
| Jeremejevite | 49 | 18 |
| Ludwigite | 82 | 52 |
| Painite | 69 | 36 |
| Rhodizite | 30 | 4 |
| Schorl | 80 | 47 |
| Vonsenite | 56 | 39 |
Mean standard deviation: ±9.5%
4. Discussion
4.1. The survival of polymers in unprotected abiotic environment
The large body of literature related to the origin of informational polymers accumulated starting from the seminal experiments by [35] and [36] has been critically reviewed [3,4] and is a matter of historical perspective [37].
The origin of (pre)genetic polymers is obscured by two major problems: the mechanism of their polymerization under plausible prebiotic conditions and their maintenance of a polymerized state outside a sufficiently protective cellular environment. Polymerization in water faces the standard-state Gibbs free energy change (ΔG°’) dilemma, essentially stating that any process entailing the release of water is, in water, thermodynamically strongly disfavoured [38]. Prebiotically plausible polymerization mechanisms were nonetheless recently proposed, based on spontaneous non-templated generation of polymers from 3’,5’-cAMP and 3’,5’-cGMP [39], or on lipid-based hydration-dehydration cycles of polymerization [40].
In order to maintain the polymerized state, the key bonds (that is, the bonds that are more susceptible to breakage) should necessarily be more stable in the polymer that in the originating monomers. In RNA the key bond is the 3’ phosphoester bond. It was noted that under defined physical-chemical conditions (namely: 60–90 °C in water) this bond is more stable by one order of magnitude in the polymer than in the monomer [15,29,42]. This observation establishes a principle but is not sufficient to account for the origin of long polymers. Did specific environments exert further protection thus allowing and/or facilitating polymer accumulation?
4.2. Why boron
Given the instability of ribose and its difficult synthesis [4], the observations that borates influence sugar chemistry [43,44,45] and their stability [11,12,13,14] has attracted the attention of origin-of-life researchers. In particular, protection of ribose by boric acid was reported [11,12]. The following aspect of boron-sugar chemistry also deserves attention. The question was posed [46,47] about the reason why the furanose and not the pyranose forms have been selected in genetic polymers. The fact that borate affords a total selection of the furanose form [11] lends further relevance to the role of boron in the prebiotic sugar world.
If nucleosides did form prebiotically by addition of sugars onto nucleic bases and not by synthesis of the sugar moiety on the bases or vice versa (which remains an unresolved problem), selective protection of certain sugars could have enhanced their accumulation under prebiotic conditions favouring the formation of nucleosides. This could in turn have kick-started (pre)genetic polymerization in a prebiotic physical-chemical frame that is not necessarily confined to Terran conditions.
We have thus explored the possibility that boron and boron-based minerals could have been potentially relevant elements in the prebiotic origin of genetic polymers. Boron exerts its protective effect on ribose [11,12,13,14] by complexing it, as such or in its nucleoside form. In order to be involved in further reactions (i.e., from ribose to nucleoside, from nucleoside to nucleotide, and from this to the RNA polymer) the sugar moiety has to be resolved from the complex with boron. The question thus applies: does a protective/enhancing function of boron exist on these down-the-line reactions?
4.3. Is boron prebiotic?
Borate minerals are abundant and numerous on Earth [48]. As for plausible prebiotic relevance of boron-based compounds, the existence of large deposits of borates in the Americas and India was reported [49]. This was taken as evidence that in prebiotic periods boric acid was present at concentrations higher than present [50]. The formation of borate salt deposits following weathering and evaporation process was discussed [50], pointing to the possible relevance, in a scenario in which boron exerts a positive protective role, of the weathering of boron minerals, and of an environment from which it is excluded resulting in its concentration. Vice versa, its sequestration in minerals would have had negative consequences. In sketching a prebiotic scenario in which a possible promoting role is played by boron, mineral weathering is a relevant aspect.
4.4. Protection of RNA by boron minerals: A rare phenomenon
We have analyzed the stability of RNA when subjected to hydrolytic attack or to attack by formamide. In these two assay conditions the average half-life of the most unstable bond (the 3’ phosphoester bond) is respectively 6.3 × 10−2 and 6.7 × 10−3 min. It was found that boron provided by boric acid (Figure 4) or by borax (data not shown) does not exert any protection, either in water or in formamide.
Borate minerals (whose stability and induced pH variation were characterized under the assay conditions) stimulate, as general trend, RNA degradation. Exception occurs in water with jeremejevite [Al6B5O15F2.5(OH)0.5], ludwigite [Mg2Fe3+BO5] and hambergite [Be2(BO3)(OH)]. These three minerals have different elemental composition, and only protect RNA at low concentration (10 µg/mL), while at higher concentration they behave like the other 17 minerals analyzed and favour degradation. Given that boron itself does not protect, that these three minerals only do so at low concentration and given their relatively long half-life in the assay conditions (see Table 2), the plausible interpretation is that protection is due to some RNA/mineral surface interaction. At concentrations higher than 10 µg/mL, release of Al and F, of Mg and Fe, and of Be, respectively, would favour degradation. At the lower 10 µg/mL concentration surface interaction with these three minerals provides RNA with a longer half-life, as reported in Figure 6 for ludwigite. Direct measurement actually showed the stable interaction between RNA and these minerals (Table 4). The fate of RNA in borates was analyzed in conjunction with formamide because of the reported formamide-based syntheses of a complete set of nucleic acid precursors [4,16] and of their trans-phosphorylation [51]. A scenario was proposed for the synthesis of (pre)genetic nucleic polymers consisting of formamide-based syntheses in the presence of the appropriate catalysts [4,42].
5. Conclusions
Boron has been proposed as a potentially relevant element in the prebiotic origin of genetic polymers, based on its known protective effect on sugars and on ribose in particular. The results of the present analysis show that boron or boron-containing minerals mostly favour a faster destabilization of the polymeric form, thus exerting a basically negative role in the polymerization scenario.
RNA does not bind very stably to any of the minerals tested and, for the minerals on which binding occurs, it is always released by pyrophosphate wash. Boron does not exert protective effect on RNA when part of a mineral, with a few interesting exceptions. Boron does not protect RNA when provided by boric acid or borax. Thus, in particular, boron does not protect RNA’s D-ribose moiety when part of the RNA chain. The reported protection by boron on ribose [11,12] are thus potentially only instrumental in the process of accumulation of the nucleic acid precursors, not of their polymerized forms, nor of their polymerization.
The specific aspect of the origin of informational polymers described here may be of general astrobiological interest. The overall structure of informational polymers and the properties of borates are not expected to vary in non-Terran environment.
Acknowledgements
This work was jointly supported by NSF and the NASA Astrobiology Program, under the NSF Center for Chemical Evolution, CHE-1004570, Italian Space Agency “MoMa project” and by ASI-INAF n. I/015/07/0 “Esplorazione del Sistema Solare”. Thanks also to Silvia Lopizzo for helpful contributions.
References
- Joyce, G.F.; Young, R.; Chang, S.; Clark, B.; Deamer, D.; DeVincenzi, D.; Ferris, J.; Irvine, W.; Kasting, J.; Kerridge, J.; Klein, H.; Knoll, A.; Walker, J. Origins of Life: The Central Concepts; Deamer, D.W., Fleischaker, G.R., Eds.; Jones & Bartlett Publishers: Boston, MA, USA, 1994. [Google Scholar]
- Gilbert, W. Origin of life: The RNA world. Nature 1986. [Google Scholar] [CrossRef]
- Orgel, L.E. The origin of life—A review of facts and speculations. Trends Biochem. Sci. 1998, 23, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Orgel, L.E. Prebiotic chemistry and the origin of the RNA world. Crit. Rev. Biochem. Mol. Biol. 2004, 39, 99–123. [Google Scholar] [CrossRef] [PubMed]
- Kruger, K.; Grabowski, P.J.; Zaug, A.J.; Sands, J.; Gottschling, D.E.; Cech, T.R. Self-splicing RNA: Autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell 1982, 31, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Guerrier-Takada, C.; Gardiner, K.; Marsh, T.; Pace, N.; Altman, S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 1983, 35, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.G.; Christensen, L.; Nielsen, P.E.; Orgel, L.E. Information transfer from DNA to peptide nucleic acids by template-directed syntheses. Nucl. Acid. Res. 1997, 25, 4792–4796. [Google Scholar] [CrossRef]
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991, 254, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Benner, S.A.; Hutter, D. Phosphates, DNA, and the search for nonterrean life: a second generation model for genetic molecules. Bioorg. Chem. 2002, 30, 62–80. [Google Scholar]
- Bean, H.D.; Anet, F.A.L.; Gould, I.R.; Hud, N.V. Glyoxylate as a backbone linkage for a prebiotic ancestor of RNA. Orig. Life Evol. Biosph. 2006, 36, 39–63. [Google Scholar] [CrossRef] [PubMed]
- Prieur, B.E. Étude de l'activité prébiotique potentielle de l'acide borique. C. R. Acad. Sci. Ser. IIC Chem. 2001, 4, 667–670. [Google Scholar]
- Ricardo, A.; Carrigan, M.A.; Olcott, A.N.; Benner, S.A. Borate minerals stabilize ribose. Science 2004. [Google Scholar] [CrossRef]
- Li, Q.; Ricardo, A.; Benner, S.A.; Winefordner, J.D.; Powell, D.H. Desorption/ionization on porous silicon mass spectrometry studies on pentose-borate complexes. Anal. Chem. 2005, 77, 4503–4508. [Google Scholar] [CrossRef] [PubMed]
- Sponer, J.E.; Sumpter, B.G.; Leszczynski, J.; Sponer, J.; Fuentes-Cabrera, M. Theoretical study on the factors controlling the stability of the borate complexes of ribose, arabinose, lyxose, and xylose. Chemistry 2008, 14, 9990–9998. [Google Scholar] [CrossRef] [PubMed]
- Ciciriello, F.; Costanzo, G.; Pino, S.; Crestini, C.; Saladino, R.; Di Mauro, E. Molecular complexity favors the evolution of ribopolymers. Biochemistry 2008, 47, 2732–2742. [Google Scholar]
- Saladino, R.; Neri, V.; Crestini, C.; Costanzo, G.; Graciotti, M.; Di Mauro, E. Synthesis and degradation of nucleic acid components by formamide and iron sulfur minerals. J. Am. Chem. Soc. 2008, 130, 15512–15518. [Google Scholar] [CrossRef] [PubMed]
- Saladino, R.; Crestini, C.; Ciciriello, F.; Pino, S.; Costanzo, G.; Di Mauro, E. From formamide to RNA: The roles of formamide and water in the evolution of chemical information. Res. Microb. 2009, 160, 441–448. [Google Scholar] [CrossRef]
- Smith, K.C.; Allen, F.W. The liberation of polynucleotides by the alkaline hydrolysis of ribonucleic acid from yeast. J. Am. Chem. Soc. 1953, 75, 2131–2133. [Google Scholar]
- Lane, B.G.; Butler, G.C. The exceptional resistance of certain oligoribonucleotides to alkaline degradation. Biochim. Biophys. Acta 1959, 33, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Kaukinen, U.; Lyytikäinen, S.; Mikkola, S.; Lönnberg, H. The reactivity of phosphodiester bonds within linear single-stranded oligoribonucleotides strongly dependent on the base sequence. Nucl. Acid. Res. 2002, 30, 468–467. [Google Scholar] [CrossRef]
- Kierzek, R. Nonenzymatic hydrolysis of oligoribonucleotides. Nucl. Acid. Res. 1992, 20, 5079–5084. [Google Scholar] [CrossRef]
- Li, Y.; Breaker, R.R. Kinetics of RNA degradation by specific base catalysis of transesterification involving the 2′-hydroxyl group. J. Am. Chem. Soc. 1999, 121, 5364–5372. [Google Scholar] [CrossRef]
- Bibillo, A.; Figlerowicz, M.; Kierzek, R. The non-enzymatic hydrolysis of oligoribonucleotides. VI. The role of biogenic polyamines. Nucl. Acid. Res. 1999, 27, 3931–3937. [Google Scholar] [CrossRef]
- Friedman, R.A.; Honig, B. A free energy analysis of nucleic acid base stacking in aqueous solution. Biophys. J. 1995, 69, 1528–1535. [Google Scholar] [CrossRef] [PubMed]
- Norberg, J.; Nilsson, L. Stacking free energy profiles for all 16 natural ribodinucleoside monophosphates in aqueous solution. J. Am. Chem. Soc. 1995, 117, 10832–10840. [Google Scholar] [CrossRef]
- Norberg, J.; Nilsson, L. A conformational free energy landscape of ApApA from molecular dynamics simulations. J. Phys. Chem. 1996, 100, 2550–2554. [Google Scholar]
- Norberg, J.; Nilsson, L. Solvent influence on base stacking. Biophys. J. 1998, 74, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.H.; Gilson, S.R.; Potter, M.J.; Gilson, M.K. The physical basis of nucleic acid base stacking. Biophys. J. 2001, 80, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Saladino, R.; Crestini, C.; Ciciriello, F.; Di Mauro, E.; Costanzo, G. Origin of informational polymers: Differential stability of phosphoester bonds in ribo monomers and oligomers. J. Biol. Chem. 2006, 281, 5790–5796. [Google Scholar] [PubMed]
- Perreault, D.M.; Anslyn, E.V. Unifying the current data on the mechanism of cleavage-transesterification of RNA. Angew. Chem. Int. Ed. Engl. 1997, 36, 432–450. [Google Scholar] [CrossRef]
- Soukup, G.; Breaker, R. Relationship between internucleotide linkage geometry and the stability of RNA. RNA 1999, 5, 1308–1325. [Google Scholar] [CrossRef] [PubMed]
- Soukup, G.A.; Breaker, RR. Nucleic acid molecular switches. Trends Biotechnol. 1999, 17, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.R.; Aures, K.; Epstein, D.J. Metal ion promoted attack of an alcohol on a phosphate diester: Modelling the role of metal ions in RNA self-splicing reactions. Chem. Comm. 1995, 23, 2431–2432. [Google Scholar] [CrossRef]
- Standard Methods for the Examination of Water and Wastewater, 19th Edition; Greenberg, A.E. (Ed.) American Public Health Association: Washington, DC, USA, 1995.
- Miller, S.L. A production of amino acids under possible primitive earth conditions. Science 1953, 117, 528–529. [Google Scholar] [CrossRef] [PubMed]
- Oró, J.; Kimball, A. Synthesis of adenine from ammonium cyanide. Biochem. Biophys. Res. Commun. 1960, 2, 407–412. [Google Scholar] [CrossRef]
- Delaye, L.; Lazcano, A. Prebiological evolution and the physics of the origin of life. Phys. Life Rev. 2005, 2, 47–64. [Google Scholar] [CrossRef] [PubMed]
- van Holde, K.E. The origin of life: A thermodynamic critique in “The origins of life and evolution”. In The origins of life and evolution; Halvorson, H.O., van Holde, K.E., Eds.; Alan R. Liss, Inc.: New York, NY, USA, 1980; pp. 31–46. [Google Scholar]
- Costanzo, G.; Pino, S.; Ciciriello, F.; Di Mauro, E. Generation of long RNA chains in water. J. Biol. Chem. 2009, 284, 33206–33216. [Google Scholar] [CrossRef] [PubMed]
- Rajamani, S.; Vlassov, A.; Benner, S.; Coombs, A.; Olasagasti, F.; Deamer, D. Lipid-assisted synthesis of RNA-like polymers from mononucleotides. Origin Life Evol. Biosph. 2008, 38, 57–74. [Google Scholar] [CrossRef]
- La Neve, P.; Altieri, F.; Fiori, M.E.; Scaloni, A.; Bozzoni, I.; Caffarelli, E. Purification, cloning, and characterization of XendoU, a novel endoribonuclease involved in processing of intron-encoded small nucleolar RNAs in Xenopus laevis. J. Biol. Chem. 2003, 278, 13026–13032. [Google Scholar] [CrossRef] [PubMed]
- Ciciriello, F.; Costanzo, G.; Pino, S.; Di Mauro, E. Spontaneous generation revisited at the molecular level. In Evolutionary Biology: Concept, Modeling and Application; Pontarotti, P., Ed.; Springer Verlag: Berlin Germany, 2009; pp. 3–22. [Google Scholar]
- Mitsuhashi, S.; Lampen, J.O. Conversion of D-xylose to D-xylulose in extracts of lactobacillus pentosus. J. Biol. Chem. 1953, 204, 1011–1018. [Google Scholar]
- Hochster, R.H.; Watson, R.W. Enzymatic isomerization of. D-xylose to D-xylulose. Arch. Biochem. Biophys. 1954, 48, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Mendicino, J. FEffect of borate on alkali-catalyzed isomerization of sugars. J. Am. Chem. Soc. 1960, 82, 4975–4979. [Google Scholar] [CrossRef]
- Beier, M.; Reck, F.; Wagner, T.; Krishnamurthy, R.; Eschenmoser, A. Chemical etiology of nucleic acid structure: Comparing pentopyranosyl-(2'→4') oligonucleotides with RNA. Science 1999, 283, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Pitsch, S.; Wendeborn, S.; Jaun, B.; Eschenmoser, A. Why pentose- and not hexose-nucleic acids? Part VII. Pyranosyl-RNA (‘p’-RNA). Helv. Chim. Acta 1993, 76, 2161–2183. [Google Scholar]
- Garrett, D.E. Borates: Handbook of Deposits, Processing, Properties, and Use; Academic Press: New York, NY, USA, 1998. [Google Scholar]
- Wells, A.F. Structural Inorganic Chemistry; Clarendon Press: Oxford, UK, 1945; p. 491. [Google Scholar]
- Kawakami, T. Tourmaline breakdown in the migmatite zone of the Ryoke metamorphic belt, SW Japan. J. Metamorph. Geol. 2001, 19, 61–75. [Google Scholar] [CrossRef]
- Costanzo, G.; Saladino, R.; Crestini, C.; Ciciriello, F.; Di Mauro, E. Nucleoside phosphorylation by phosphate minerals. J. Biol. Chem. 2007, 282, 16729–16735. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Cossetti, C.; Crestini, C.; Saladino, R.; Mauro, E.D. Borate Minerals and RNA Stability. Polymers 2010, 2, 211-228. https://doi.org/10.3390/polym2030211
AMA Style
Cossetti C, Crestini C, Saladino R, Mauro ED. Borate Minerals and RNA Stability. Polymers. 2010; 2(3):211-228. https://doi.org/10.3390/polym2030211
Chicago/Turabian StyleCossetti, Cristina, Claudia Crestini, Raffaele Saladino, and Ernesto Di Mauro. 2010. "Borate Minerals and RNA Stability" Polymers 2, no. 3: 211-228. https://doi.org/10.3390/polym2030211