
Novel Correlations between Spectroscopic and Morphological Properties of Activated Carbons from Waste Coffee Grounds


 ,
,  ,
,  ,
,  ,
,  , and
, and
Abstract
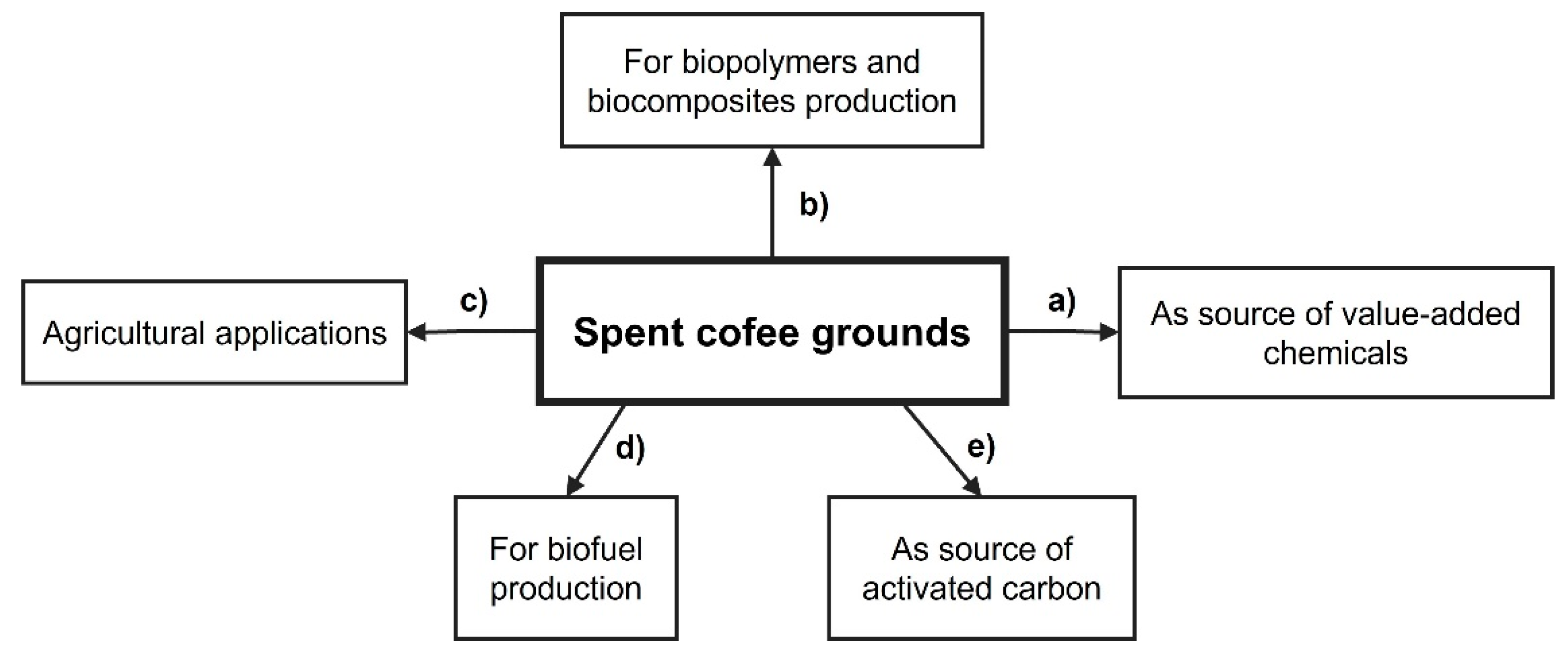
:1. Introduction
2. Materials and Methods
2.1. Raw Materials
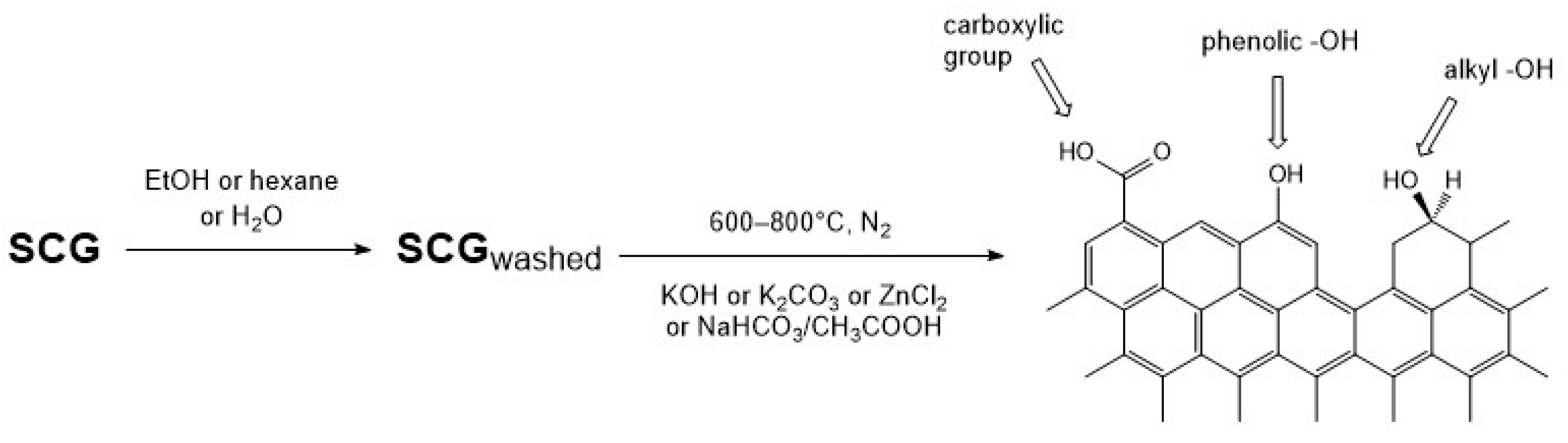
2.2. Preparation of Activated Carbons
2.3. Textural and Chemical Characterization
3. Results and Discussion
3.1. Characterization of the Starting Spent Coffee Grounds
3.2. Thermal Behavior of Raw Materials A and R and of the A,R/KOH 1/1 Mixtures
3.3. Chemical Characterization of the Activated Carbons
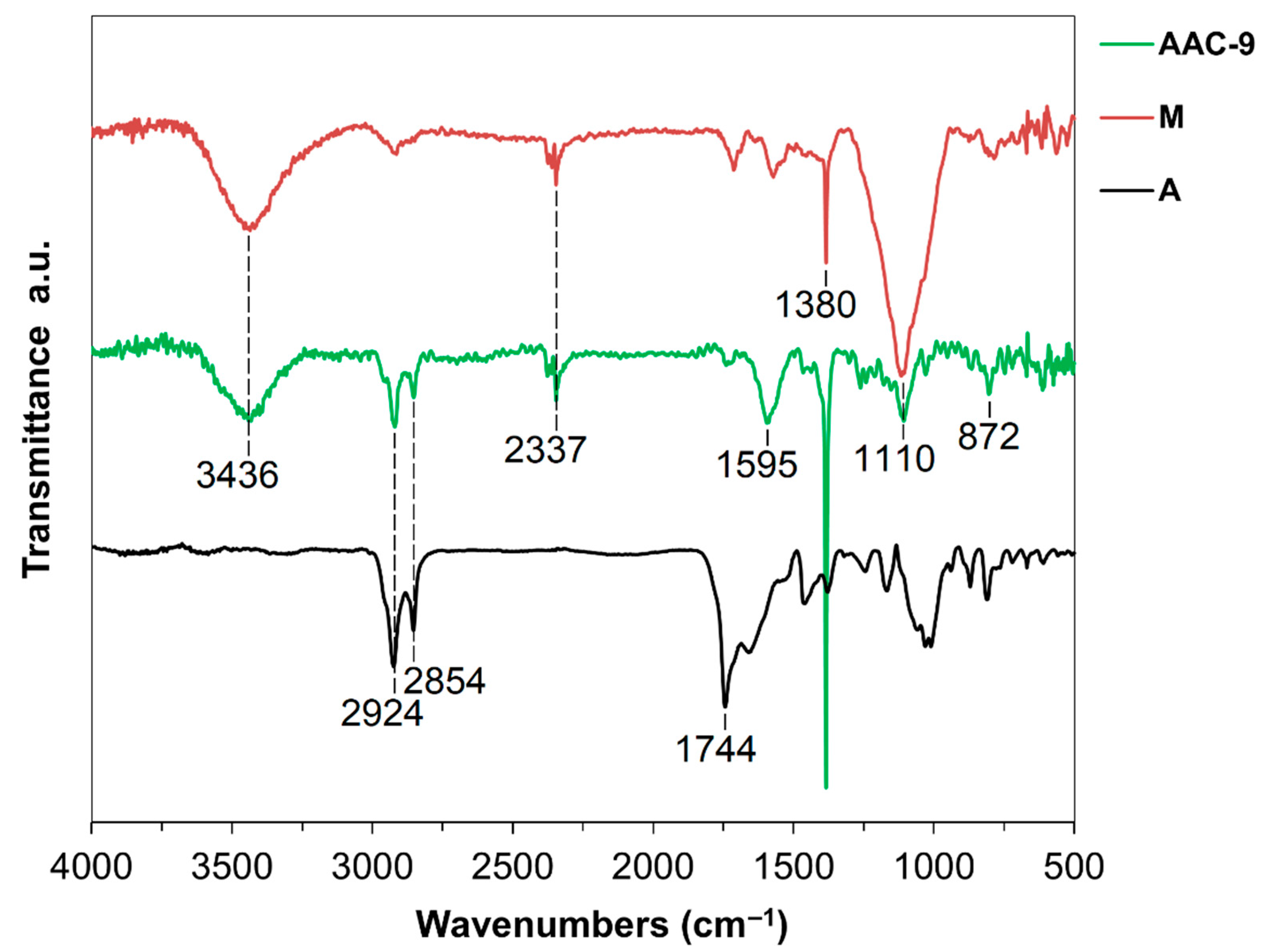
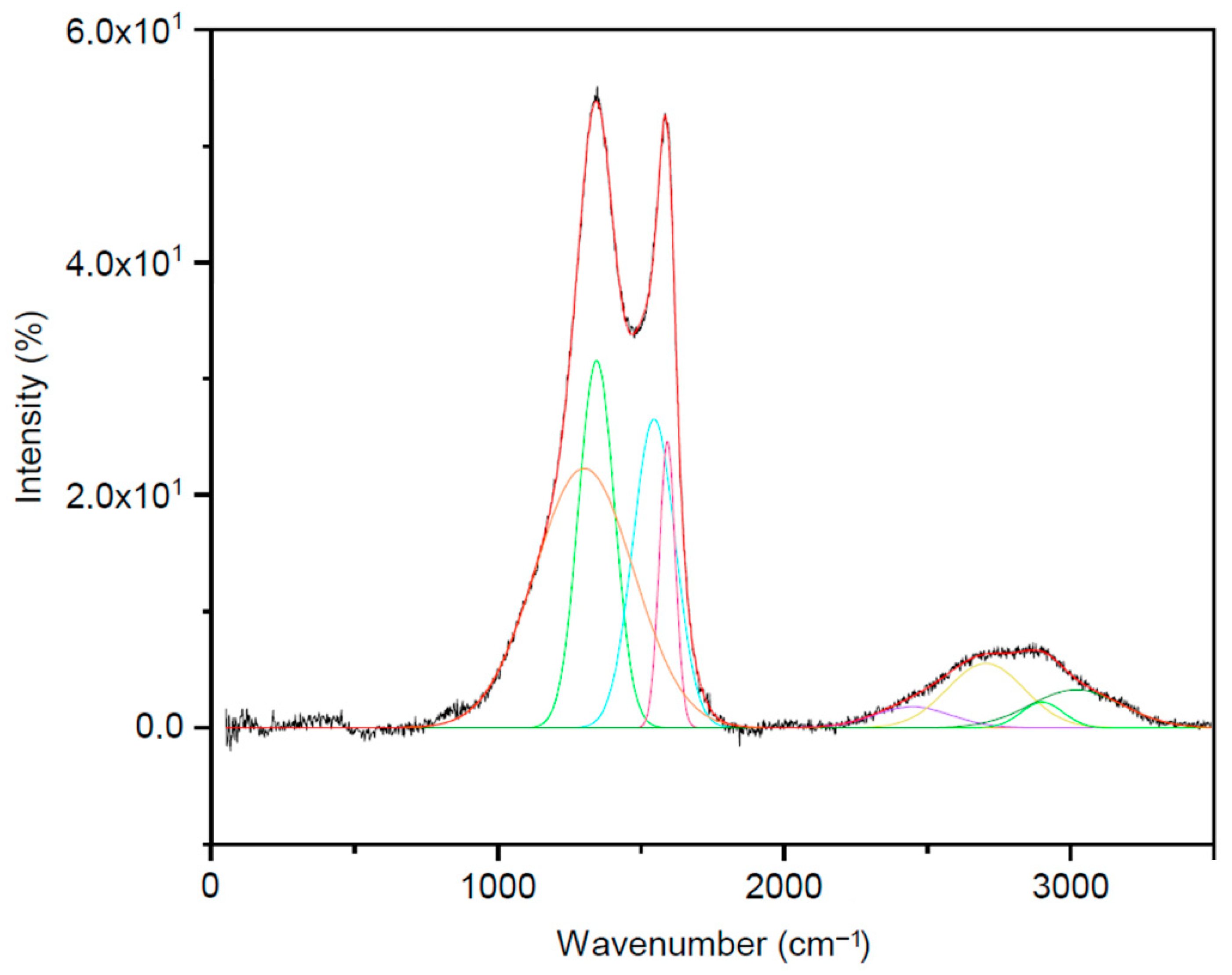
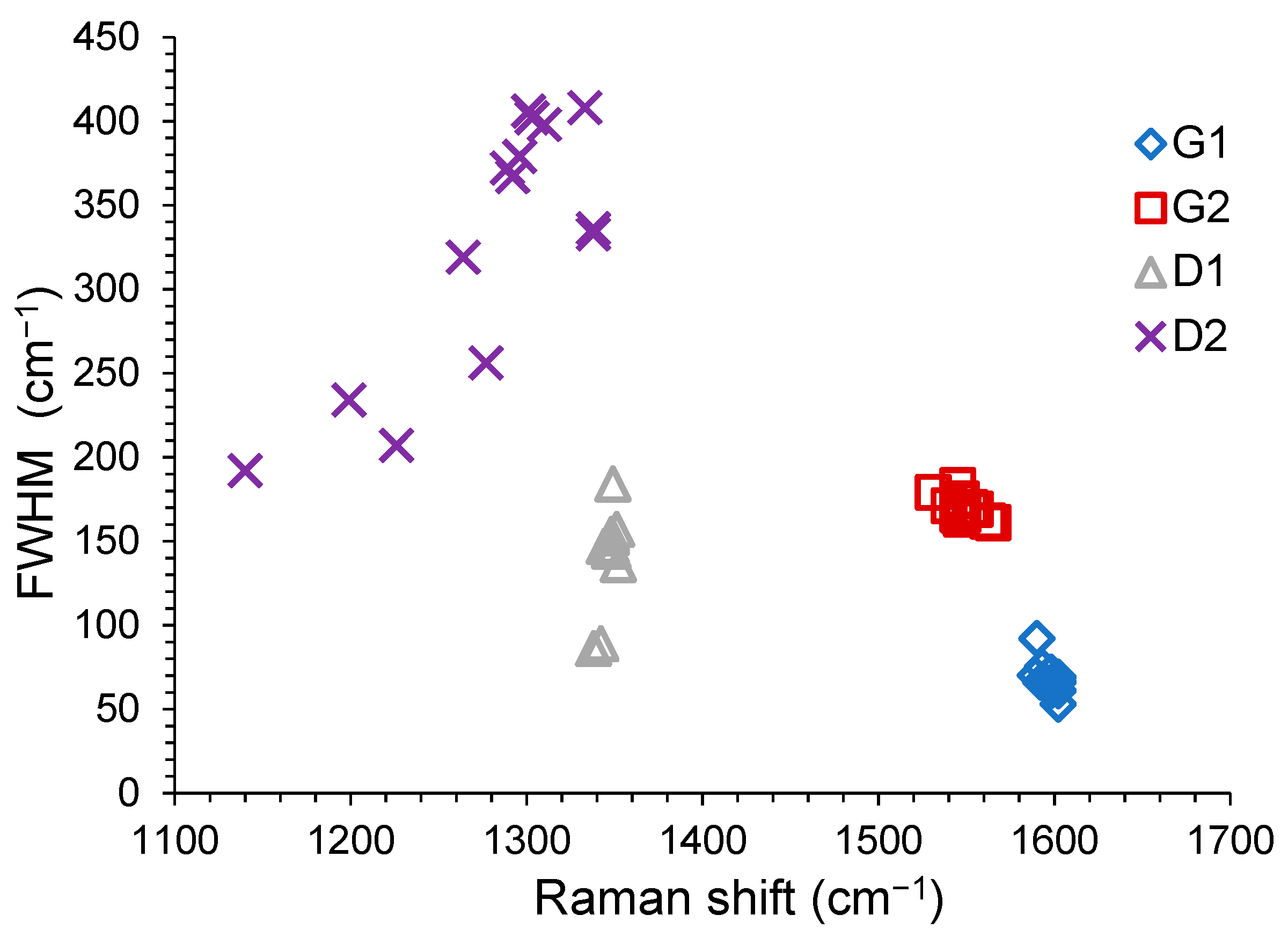
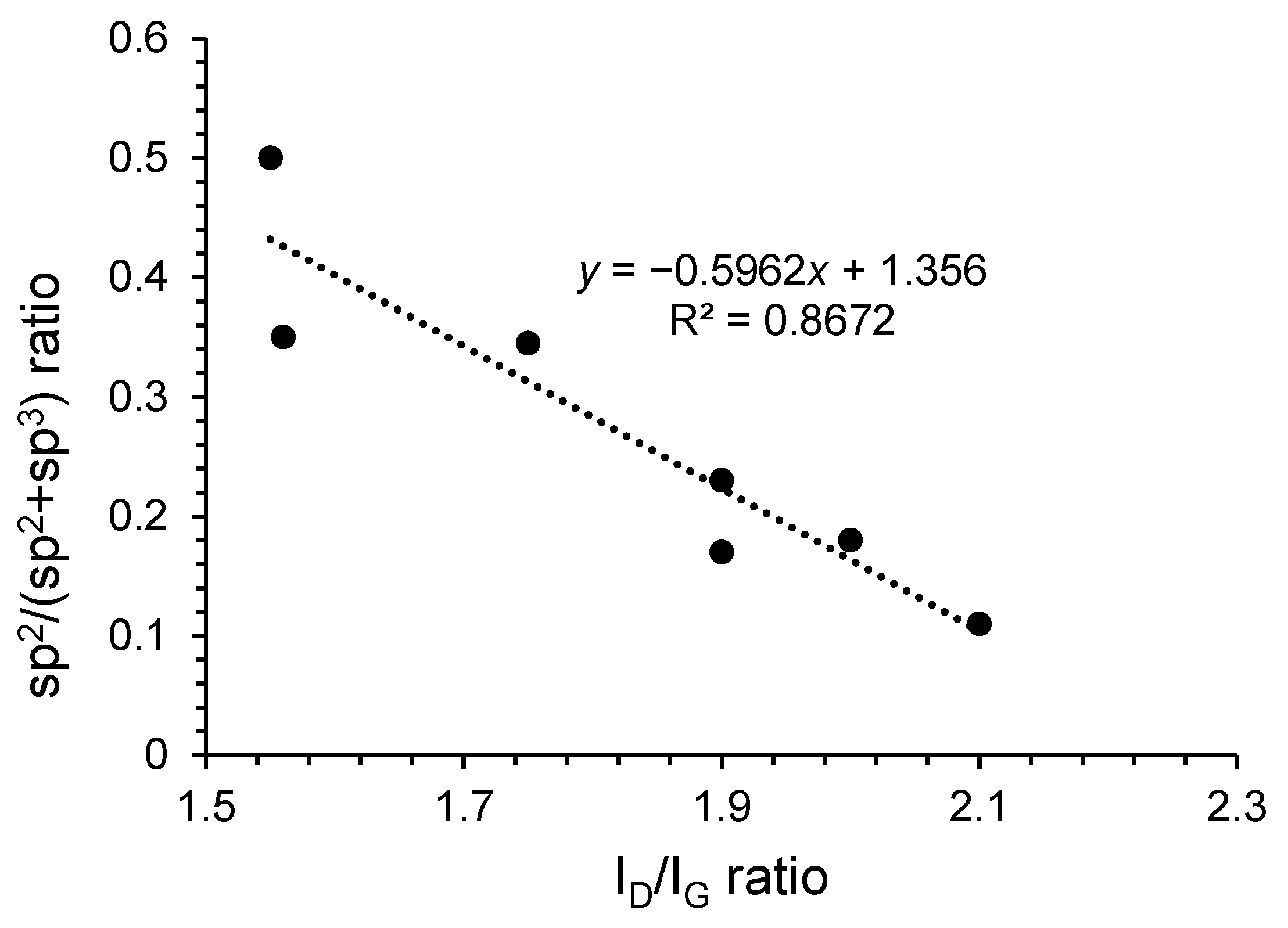
3.3.1. Fourier Transform Infrared (FT-IR) and Raman Data
- (a)
- the G1 position is quite stable: for RACs in the range 1589–1599 cm−1 (FWHM of 66.5 ± 3.5 cm−1) and for AACs in the range 1592–1603 cm−1 (FWHM of 68 ± 7 cm−1);
- (b)
- the D1 position is quite stable: for RACs in the range 1342–1352 cm−1 (FWHM of 140.5 ± 13.5 cm−1) and for AACs in the range 1344–1354 cm−1 (FWHM of 164 ± 20 cm−1);
- (c)
- the G2 position is less stable and the signals are broader: for RACs in the range 1531–1555 cm−1 (FWHM of 174.5 ± 4.5 cm−1) and for AACs in the range 1540–1565 cm−1 (FWHM of 172 ± 11 cm−1);
- (d)
- the D2 position is significantly less stable and the signals are broader: for RACs in the range 1140–1350 cm−1 (FWHM of 272 ± 80 cm−1) and for AACs in the range 1199–1333 cm−1 (FWHM of 321 ± 87 cm−1).
3.3.2. X-ray Diffraction (XRD) and X-ray Photoelectron Spectroscopy (XPS)
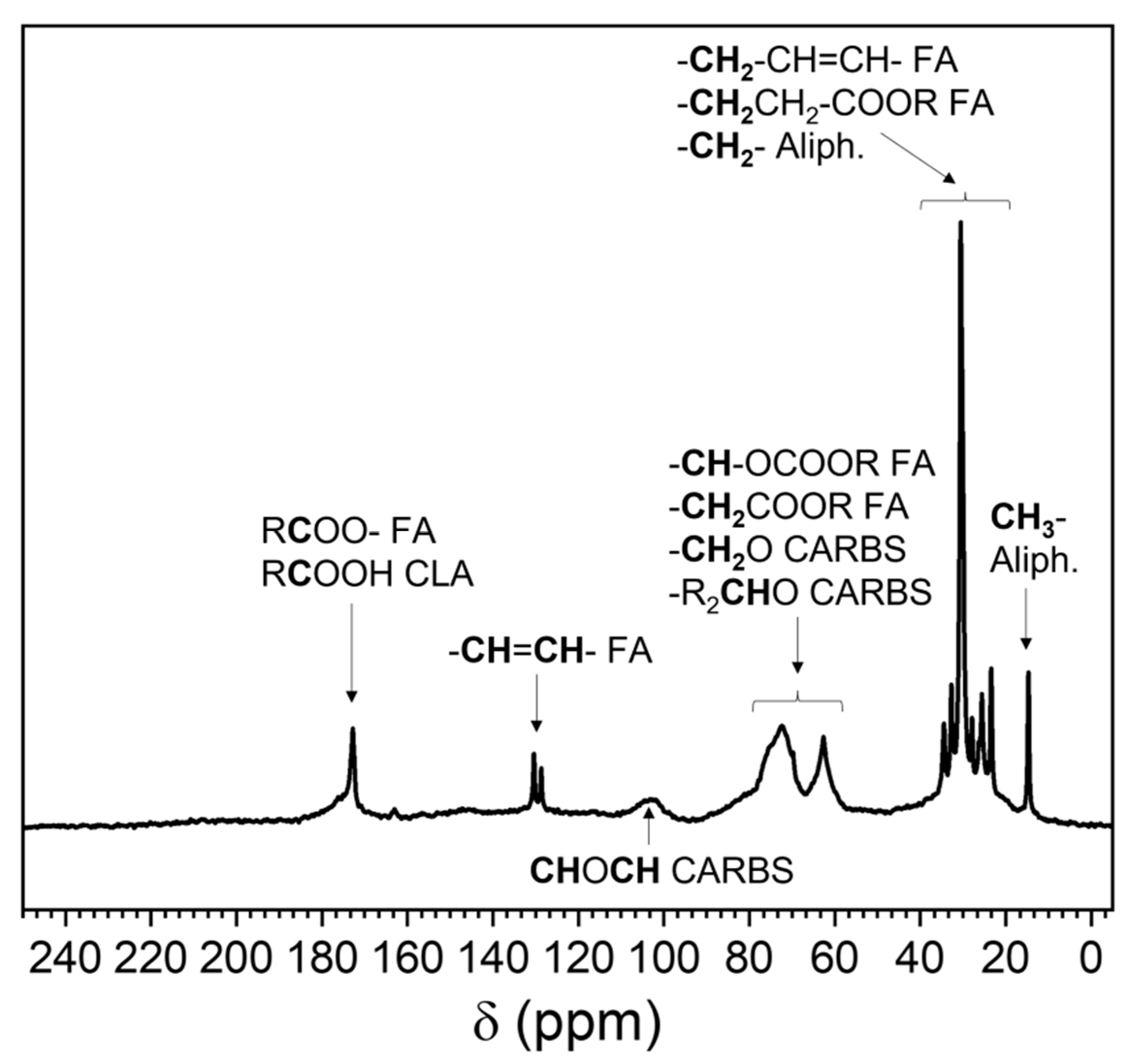
3.3.3. 13C Nuclear Magnetic Resonance (NMR)
3.3.4. Surface Chemistry Characterization
3.4. Physical and Textural Characterization
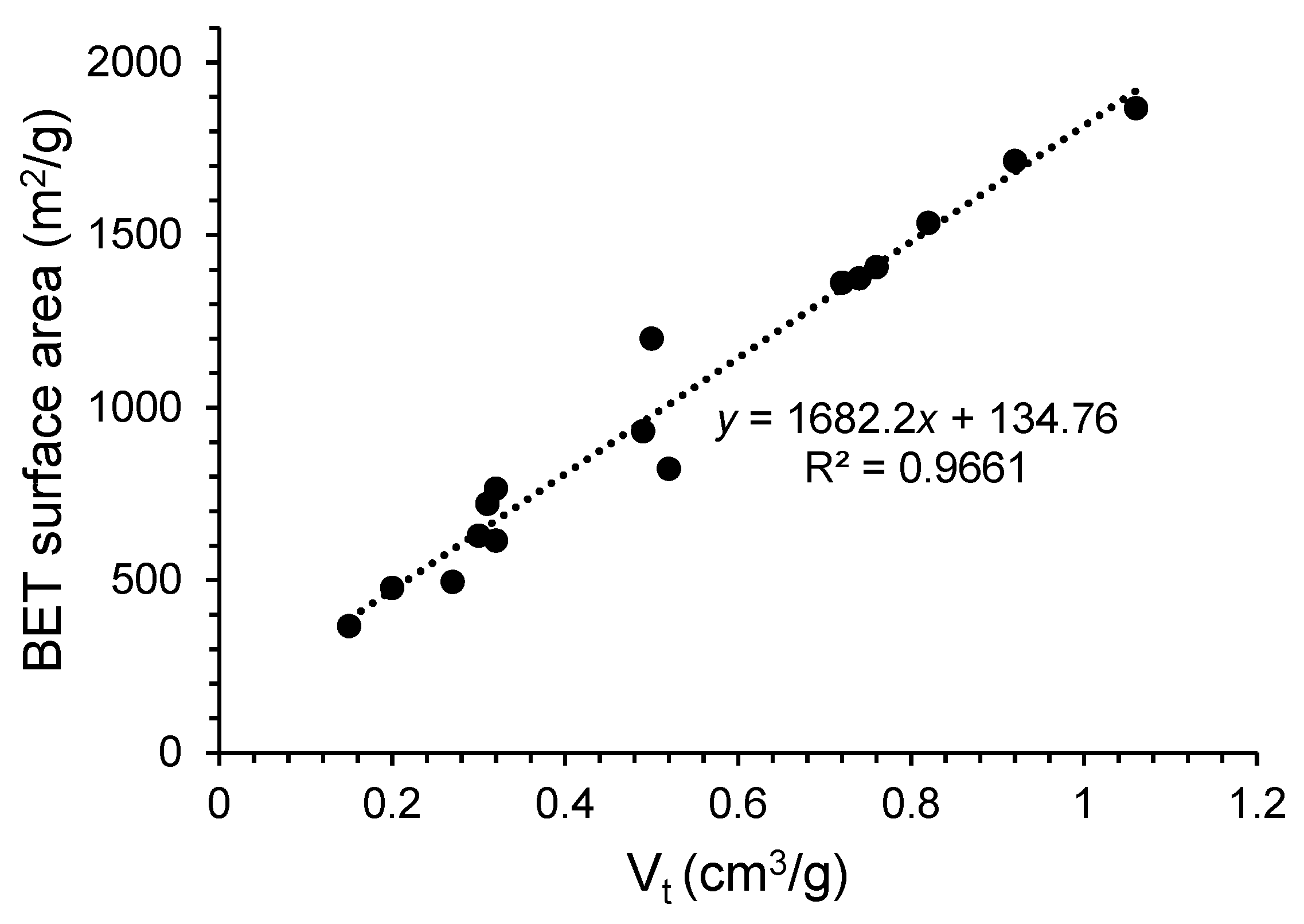
3.4.1. Brunauer–Emmett–Teller (BET) Analysis and Pore Size Distribution
3.4.2. Environmental Scanning Electron Microscopy (ESEM) and Transmission Electron Microscopy (TEM)
3.5. General Considerations on Characterization Data and Correlations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Galanakis, C. Handbook of Coffee Processing By-Products: Sustainable Applications, 1st ed.; Academic Press: London, UK, 2017; Chapters 3 and 9, In Particular. [Google Scholar]
- Zabaniotou, A.; Kamaterou, P. Food waste valorisation advocating circular bioeconomy. A critical review of potentialities and perspectives of spent coffee grounds biorefinery. J. Clean. Prod. 2019, 211, 1553–1566. [Google Scholar] [CrossRef]
- Campos-Vega, R.; Loarca-Pina, G.; Vergara-Castaneda, H.A.; Oomah, B.D. Spent coffee grounds: A review on current research and future prospects. Trends Food Sci. Technol. 2015, 45, 24–36. [Google Scholar] [CrossRef]
- Karmee, S.K. A spent coffee grounds based biorefinery for the production of biofuels, biopolymers, antioxidants and biocomposites. Waste Manag. 2018, 72, 240–254. [Google Scholar] [CrossRef]
- Osman, A.I.; Abdelkader, A.; Farrell, C.; Rooney, D.; Morgan, K. Reusing, recycling and up-cycling of biomass: A review of practical and kinetic modelling approaches. Fuel Process. Technol. 2019, 192, 179–202. [Google Scholar] [CrossRef]
- Atabani, A.E.; Al-Muhtaseb, A.H.; Kumar, G.; Saratale, G.D.; Aslam, M.; Khan, H.A.; Said, Z.; Mahmoud, E. Valorization of spent coffee grounds into biofuels and value-added products: Pathway towards integrated bio-refinery. Fuel 2019, 254, 115640. [Google Scholar] [CrossRef]
- Massaya, J.; Pereira, A.P.; Mills-Lamptey, B.; Benjamin, J.; Chuck, C.J. Conceptualization of a spent coffee grounds biorefinery: A review of existing valorisation approaches. Food Bioprod. Process. 2019, 118, 149–166. [Google Scholar] [CrossRef]
- Mussatto, S.I.; Machado, E.M.S.; Martins, S.; Teixeira, J.A. Production, composition, and application of coffee and its industrial residues. Food Bioprocess Technol. 2011, 4, 661–672. [Google Scholar] [CrossRef] [Green Version]
- Girotto, F.; Pivato, A.; Cossu, R.; Nkeng, G.E.; Lavagnolo, M.C. The broad spectrum of possibilities for spent coffee grounds valorisation. J. Mater. Cycles Waste Manag. 2018, 20, 695–701. [Google Scholar] [CrossRef]
- Stylianou, M.; Agapiou, A.; Omirou, M.; Vyrides, I.; Ioannides, I.M.; Maratheftis, G.; Fasoula, D. Converting environmental risks to benefits by using spent coffee grounds (SCG) as a valuable resource. Environ. Sci. Pollut. Res. 2018, 25, 35776–35790. [Google Scholar] [CrossRef]
- Peshev, D.; Mitev, D.; Peeva, L.; Peev, G. Valorization of spent coffee grounds—A new approach. Sep. Purif. Technol. 2018, 192, 271–277. [Google Scholar] [CrossRef]
- Nguyen, Q.A.; Cho, E.J.; Lee, D.S.; Bae, H.J. Development of an advanced integrative process to create valuable biosugars including manno-oligosaccharides and mannose from spent coffee grounds. Bioresour. Technol. 2019, 272, 209–216. [Google Scholar] [CrossRef]
- Tongcumpou, C.; Usapein, P.; Tuntiwiwattanapun, N. Complete utilization of wet spent coffee grounds waste as a novel feedstock for antioxidant, biodiesel, and bio-char production. Ind. Crop. Prod. 2019, 138, 111484. [Google Scholar] [CrossRef]
- Dattatraya Saratale, G.; Bhosale, R.; Shobana, S.; Banu, J.R.; Pugazhendhi, A.; Mahmoud, E.; Sirohi, R.; Bhatia, S.K.; Atabani, A.E.; Mulone, V.; et al. A review on valorization of spent coffee grounds (SCG) towards biopolymers and biocatalysts production. Bioresour. Technol. 2020, 314, 123800. [Google Scholar] [CrossRef]
- Tan, M.Y.; Kuan, H.T.N.; Lee, M.C. Characterization of alkaline treatment and fiber content on the physical, thermal and mechanical properties of ground coffee waste/oxobiodegradable HDPE biocomposites. Int. J. Polym. Sci. 2017, 2017, 6258151. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.; Chang, S.J.; Wi, S.; Kim, S. Spent coffee grounds as supporting materials to produce biocomposite PCM with natural waxes. Chemosphere 2019, 235, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.; Ko, Y.G.; Do, T.; Jung, Y.; Kim, S.W.; Choi, U.S. Spent coffee grounds: Massively supplied carbohydrate polymer applicable to electrorheology. Colloids Surfaces A Physicochem. Eng. Asp. 2019, 562, 392–401. [Google Scholar] [CrossRef]
- Cruz, R.; Mendes, E.; Torrinha, A.; Morais, S.; Pereira, J.A.; Baptista, P.; Casal, S. Revalorization of spent coffee residues by a direct agronomic approach. Food Res. Int. 2015, 71, 190–196. [Google Scholar] [CrossRef]
- Elbl, J.; Plosek, L.; Kintl, A.; Prichystalova, J.; Zahora, J.; Friedel, J.K. The effect of increased doses of compost on leaching of mineral nitrogen from arable land. Pol. J. Environ. Stud. 2014, 23, 697–703. [Google Scholar]
- Sikka, S.S.; Chawla, I.S. Effect of feeding spent coffee grounds on the feedlot performance of carcass quality of fattening pigs. Agric. Wastes 1986, 18, 305–308. [Google Scholar] [CrossRef]
- Mendoza Martinez, C.L.; Alves Rocha, E.P.; Oliveira Carneiro, A.C.; Borges Gomes, F.J.; Ribas Batalha, L.A.; Vakkilainen, E.; Cardoso, M. Characterization of residual biomasses from the coffee production chain and assessment the potential for energy purposes. Biomass Bioenerg. 2019, 120, 68–76. [Google Scholar] [CrossRef]
- Nguyen, H.C.; Nguyen, M.L.; Wang, F.M.; Juan, H.Y.; Su, C.H. Biodiesel production by direct transesterification of wet spent coffee grounds using switchable solvent as a catalyst and solvent. Bioresour. Technol. 2020, 296, 122334. [Google Scholar] [CrossRef]
- Dohlert, P.; Weidauer, M.; Enthaler, S. Spent coffee ground as source for hydrocarbon fuels. J. Energy Chem. 2016, 25, 146–152. [Google Scholar] [CrossRef]
- Fischer, A.; Du, S.; Valla, J.A.; Bollas, G.M. The effect of temperature, heating rate and ZSM-5 catalyst on the product selectivity of the fast pyrolysis of spent coffee grounds. RCS Adv. 2015, 5, 29252–29261. [Google Scholar] [CrossRef]
- Tehrani, N.F.; Aznar, J.S.; Kiros, Y. Coffee extract residue for production of ethanol and activated carbons. J. Clean. Prod. 2015, 91, 64–70. [Google Scholar] [CrossRef]
- Vardon, D.R.; Moser, B.R.; Zheng, W.; Witkin, K.; Evangelista, R.L.; Strathmann, T.J.; Rajagopalan, K.; Sharma, B.K. Complete utilization of spent coffee grounds to produce biodiesel, bio-oil and biochar. ACS Sustain. Chem. Eng. 2013, 1, 1286–1294. [Google Scholar] [CrossRef]
- Primaz, C.T.; Schena, T.; Lazzari, E.; Caramao, E.B.; Jaques, R.A. Influence of the temperature in the yield and composition of the bio-oil from the pyrolysis of spent coffee grounds: Characterization by comprehensive two dimensional gas chromatography. Fuel 2018, 232, 572–580. [Google Scholar] [CrossRef]
- Cay, H.; Duman, G.; Yanik, J. Two-step gasification of biochar for hydrogen-rich gas production: Effect of the biochar type and catalyst. Energy Fuels 2019, 33, 7398–7405. [Google Scholar] [CrossRef]
- Jutakridsada, P.; Prajaksud, C.; Kuboonya-Aruk, L.; Theerakulpisut, S.; Kamwilaisak, K. Adsorption characteristics of activated carbon prepared from spent ground coffee. Clean Technol. Environ. Policy 2016, 18, 639–645. [Google Scholar] [CrossRef]
- Alcaraz, L.; Escudero, M.E.; Alguacil, F.J.; Llorente, I.; Urbieta, A.; Fernandez, P.; Lopez, F.A. Dysprosium removal from water using active carbons obtained from spent coffee ground. Nanomaterials 2019, 9, 1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babu, N.A.; Reddy, D.S.; Kumar, G.S.; Ravindhranath, K.; Mohan, G.V.K. Removal of lead and fluoride from contaminated water using exhausted coffee grounds based bio-sorbent. J. Environ. Manag. 2018, 218, 602–612. [Google Scholar] [CrossRef]
- Fonseca Alves, A.C.; Antero, R.V.P.; de Oliveira, S.B.; Ojala, S.A.; Scalize, P.S. Activated carbon produced from waste coffee grounds for an effective removal of bisphenol-A in aqueous medium. Environ. Sci. Pollut. Res. 2019, 26, 24850–24862. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.F.; Wu, C.L.; Kuo, S.W.; Hung, W.S.; Lee, K.J.; Tsai, H.C.; Chang, C.J.; Lai, J.Y. Preparation of efficient photothermal materials from waste coffee grounds for solar evaporation and water purification. Sci. Rep. 2020, 10, 12769. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Liang, C. Evaluation of the engineering properties of powdered activated carbon amendments in porous asphalt pavement. Processes 2021, 9, 582. [Google Scholar] [CrossRef]
- Suganya, S.; Kumar, P.S. Influence of ultrasonic waves on preparation of active carbon from coffee waste for the reclamation of effluents containing Cr(VI) ions. J. Ind. Eng. Chem. 2018, 60, 418–430. [Google Scholar] [CrossRef]
- Rovani, S.; Censi, M.T.; Pedrotti, S.L., Jr.; Lima, E.C.; Cataluna, R.; Fernandes, A.N. Development of a new adsorbent from agro-industrial waste and its potential use in endocrine disruptor compound removal. J. Hazard. Mater. 2014, 271, 311–320. [Google Scholar] [CrossRef]
- Nowicki, P.; Skibiszewska, P.; Pietrzak, R. Hydrogen sulphide removal on carbonaceous adsorbents prepared from coffee industry waste materials. Chem. Eng. J. 2014, 248, 208–215. [Google Scholar] [CrossRef]
- Querejeta, N.; Gil, M.V.; Rubiera, F.; Pevida, C. Sustainable coffee-based CO2 adsorbents: Toward a greener production via hydrothermal carbonization. Greenh. Gases Sci. Technol. 2018, 8, 309–323. [Google Scholar] [CrossRef]
- Travis, W.; Gadipelli, S.; Guo, Z. Superior CO2 adsorption from waste coffee ground derived carbons. RCS Adv. 2015, 5, 29558–29562. [Google Scholar] [CrossRef]
- Mohamed, M.G.; Ahmed, M.M.M.; Du, W.T.; Kuo, S.W. Meso/Microporous carbon from conjugated hyper-crosslinked polymers based on tetraphenylethene for high-performance CO2 capture and supercapacitor. Molecules 2021, 16, 738. [Google Scholar] [CrossRef]
- Samy, M.M.; Mohamed, M.G.; Kuo, S.W. Directly synthesized nitrogen and oxygen doped microporous carbons derived from a bio-derived polybenzoxazine exhibiting high-performance supercapacitance and CO2 uptake. Eur. Polym. J. 2020, 138, 109954. [Google Scholar] [CrossRef]
- Kemp, K.C.; Baek, S.B.; Lee, W.G.; Meyyappan, M.; Kim, K.S. Activated carbon derived from waste coffee grounds for stable methane storage. Nanotechnology 2015, 26, 385602–385609. [Google Scholar] [CrossRef] [PubMed]
- Akasaka, H.; Takahata, T.; Toda, I.; Ono, H.; Ohshio, S.; Himeno, S.; Kokubu, T.; Saitoh, H. Hydrogen storage ability of porous carbon material fabricated from coffee bean wastes. Int. J. Hydrogen Energy 2011, 36, 580–585. [Google Scholar] [CrossRef]
- Um, J.H.; Kim, Y.; Ahn, C.Y.; Kim, J.; Sung, Y.E.; Cho, Y.H.; Kim, S.S.; Yoon, W.S. Biomass waste, coffee grounds-derived carbon for lithium storage. J. Electrochem. Sci. Technol. 2018, 9, 163–168. [Google Scholar] [CrossRef]
- Oh, W.D.; Lisak, G.; Webster, R.D.; Liang, Y.N.; Veksha, A.; Giannis, A.; Moo, J.G.S.; Lim, J.W.; Lim, T.T. Insights into the thermolytic transformation of lignocellulosic biomass waste to redox-active carbocatalyst: Durability of surface active sites. Appl. Catal. B Environ. 2018, 233, 120–129. [Google Scholar] [CrossRef]
- Goncalves, M.; Guerreiro, M.C.; Alves de Oliveira, L.C.; Soares de Castro, C. A friendly environmental material: Iron oxide dispersed over activated carbon from coffee husk for organic pollutants removal. J. Environ. Manag. 2013, 127, 206–211. [Google Scholar] [CrossRef]
- Hung, Y.H.; Liu, T.Y.; Chen, H.Y. Renewable coffee waste-derived porous carbons as anode materials for high-performance sustainable microbial fuel cells. ACS Sustain. Chem. Eng. 2019, 7, 16991–16999. [Google Scholar] [CrossRef]
- Lee, M.E.; Kwak, H.W.; Jin, H.J.; Yun, Y.S. Waste beverage coffee-induced hard carbon granules for sodium-ion batteries. ACS Sustain. Chem. Eng. 2019, 7, 12734–12740. [Google Scholar] [CrossRef]
- Zhao, P.; Aboonasr Shiraz, M.H.; Zhu, H.; Liu, Y.; Tao, L.; Liu, J. Hierarchically porous carbon from waste coffee grounds for high-performance Li-Se batteries. Electrochim. Acta 2019, 325, 134931–134938. [Google Scholar] [CrossRef]
- Choi, J.; Zequine, C.; Bhoyate, S.; Lin, W.; Li, X.; Kahol, P.; Gupta, R. Waste coffee management: Deriving high-performance supercapacitors using nitrogen-doped coffee-derived carbon. C J. Carbon Res. 2019, 5, 44. [Google Scholar] [CrossRef] [Green Version]
- Frohlich, A.C.; dos Reis, G.S.; Pavan, F.A.; Lima, E.C.; Foletto, E.L.; Dotto, G.L. Improvement of activated carbon characteristics by sonication and its application for pharmaceutical contaminant adsorption. Environ. Sci. Pollut. Res. 2018, 25, 24713–24725. [Google Scholar] [CrossRef]
- Kim, Y.; Lee, J.; Yi, H.; Tsang, Y.F.; Kwon, E.E. Investigation into role of CO2 in two-stage pyrolysis of spent coffee grounds. Bioresour. Technol. 2019, 272, 48–53. [Google Scholar] [CrossRef]
- Laksaci, H.; Khelifi, A.; Trari, M.; Addoun, A. Synthesis and characterization of microporous activated carbon from coffee grounds using potassium hydroxide. J. Clean. Prod. 2017, 147, 254–262. [Google Scholar] [CrossRef]
- Laksaci, H.; Khelifi, A.; Belhamdi, B.; Trari, M. Valorization of coffee grounds into activated carbon using physic-chemical activation by KOH/CO2. J. Environ. Chem. Eng. 2017, 5, 5061–5066. [Google Scholar] [CrossRef]
- Rodriguez-Reinoso, F.; Molina-Sabio, M.; Gonzalez, M.T. The use of steam and CO2 as activating agents in the preparation of activated carbons. Carbon 1995, 33, 15–23. [Google Scholar] [CrossRef]
- Chiu, Y.H.; Lin, L.Y. Effect of activating agents for producing activated carbon using a facile one-step synthesis with waste coffee grounds for symmetric supercapacitors. J. Taiwan Inst. Chem. Eng. 2019, 101, 177–185. [Google Scholar] [CrossRef]
- Wang, C.H.; Wen, W.C.; Hsu, H.C.; Yao, B.Y. High-capacitance KOH-activated nitrogen-containing porous carbon material from waste coffee grounds in supercapacitors. Adv. Power Technol. 2016, 27, 1387–1395. [Google Scholar] [CrossRef]
- Pujol, D.; Liu, C.; Gominho, J.; Olivella, M.A.; Fiol, N.; Villaescusa, I.; Pereira, H. The chemical composition of exhausted coffee waste. Ind. Crop. Prod. 2013, 50, 423–429. [Google Scholar] [CrossRef]
- Wilson, L.; Yang, W.; Blasiak, W.; John, G.R.; Mhilu, C.F. Thermal characterization of tropical biomass feedstocks. Energy Conv. Manag. 2011, 52, 191–198. [Google Scholar] [CrossRef]
- Pawlicka, A.; Doczekalska, B. Determination of surface oxygen functional groups of active carbons according to Bohem’s titration method. For. Wood Technol. 2013, 84, 11–14. [Google Scholar]
- Kumar, A.; Prasad, B.; Mishra, I.M. Adsorptive removal of acrylonitrile by commercial grade activated carbon: Kinetics, equilibrium and thermodynamics. J. Hazard. Mater. 2007, 152, 589–600. [Google Scholar] [CrossRef]
- Ballesteros, L.F.; Teixeira, J.A.; Mussatto, S.I. Chemical, functional and structural properties of spent coffee grounds and coffee silverskin. Food Bioprocess Technol. 2014, 7, 3493–3503. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.K.; Hajaligol, M.R.; Martoglio Smith, P.A.; Wooten, J.B.; Baliga, V. Characterization of char pyrolysis of chlorogenic acid. Energy Fuels 2000, 14, 1083–1093. [Google Scholar] [CrossRef]
- Wang, N.; Lim, L.T. Fourier transform infrared and physicochemical analyses of roasted coffee. J. Agric. Food Chem. 2012, 60, 5446–5453. [Google Scholar] [CrossRef]
- Li, X.; Strezov, V.; Kan, T. Energy recovery potential analysis of spent coffee grounds pyrolysis products. J. Anal. Appl. Pyrolysis 2014, 110, 79–87. [Google Scholar] [CrossRef]
- Skreiberg, A.; Skreiberg, O.; Sandquist, J.; Sorum, L. TGA and macro-TGA characterization of biomass fuels and fuel mixtures. Fuel 2011, 90, 2182–2197. [Google Scholar] [CrossRef]
- Fermoso, J.; Masek, O. Thermochemical decomposition of coffee ground residues by TG-MS: A kinetic study. J. Anal. Appl. Pyrolysis 2018, 130, 358–367. [Google Scholar] [CrossRef] [Green Version]
- Chia, C.H.; Gong, B.; Joseph, S.D.; Marjo, C.E.; Munroe, P.; Rich, A.M. Imaging of mineral-enriched biochar by FTIR, Raman and SEM-EDX. Vib. Spectrosc. 2012, 62, 248–257. [Google Scholar] [CrossRef]
- Centrone, A.; Brambilla, L.; Renouard, T.; Gherghel, L.; Mathis, C.; Mullen, K.; Zerbi, G. Structure of new carbonaceous materials: The role of vibrational spectroscopy. Carbon 2005, 43, 1593–1609. [Google Scholar] [CrossRef]
- Zhuo, Y.; Lemaignen, L.; Chatzakis, I.N.; Reed, G.P.; Dugwell, D.R.; Kandiyoti, R. An attempt to correlate conversion in pyrolysis and gasification with FT-IR spectra of coals. Energy Fuels 2000, 14, 1049–1058. [Google Scholar] [CrossRef]
- Shimodaira, N.; Masui, A. Raman spectroscopic investigations of activated carbon materials. J. Appl. Phys. 2002, 92, 902–909. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Robertson, J. Interpretation of Raman spectra of disordered and amorphous carbon. Phys. Rev. B 2000, 61, 14095–14107. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, A.C. Raman spectroscopy of graphene and graphite: Disorder, electron-phonon coupling, doping and nonadiabatic effects. Solid State Commun. 2007, 143, 47. [Google Scholar] [CrossRef]
- Kaniyoor, A.; Ramaprabhu, S. A Raman spectroscopic investigation of graphite oxide derived graphene. AIP Adv. 2012, 2, 032183. [Google Scholar] [CrossRef] [Green Version]
- Malard, L.M.; Pimenta, M.A.; Dresselhaus, G.; Dresselhaus, M.S. Raman spectroscopy in graphene. Phys. Rep. 2008, 473, 51–87. [Google Scholar] [CrossRef]
- Pimenta, M.A.; Dresselhaus, G.; Dresselhaus, M.S.; Cancado, L.G.; Jorio, A.; Saito, R. Studying disorder in graphite-based systems by Raman spectroscopy. Phys. Chem. Chem. Phys. 2007, 9, 1276–1291. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.H.; Deng, Y.M.; Zhao, Q. Temperature-dependent Raman spectra and anomalous Raman phenomenon of highly oriented pyrolytic graphite. Phys. Rev. 1998, 58, 5435–5439. [Google Scholar] [CrossRef] [Green Version]
- Morgan, D.J. Comments on the XPS analysis of carbon materials. J. Carbon Res. 2021, 7, 51. [Google Scholar] [CrossRef]
- Lesiak, B.; Kover, L.; Toth, J.; Zemek, J.; Jiricek, P.; Kromba, A.; Rangam, N. C sp2/sp3 hybridisations in carbon materials—XPS and (X)AES study. Appl. Surf. Sci. 2018, 452, 223–231. [Google Scholar] [CrossRef]
- Li, X.; Kang, Y.; Haghighi, M. Investigation of pore size distributions of coals with different structures by nuclear magnetic resonance (NMR) and mercury intrusion porosimetry. Measurement 2018, 116, 122–128. [Google Scholar] [CrossRef]
- McBeath, A.V.; Smernik, R.J.; Krull, E.S.; Lehmann, J. The influence of feedstock and production temperature on biochar carbon chemistry: A solid-state 13C NMR study. Biomass Bioenerg. 2014, 60, 121–129. [Google Scholar] [CrossRef]
- Chen, L.S.; Wang, L.I.; Pan, T.Y.; Zhang, Y.Y.; Zhang, D.X. Calibration of solid state NMR carbon structural parameters and application in coal structure analysis. J. Fuel Chem. Technol. 2017, 45, 1153–1163. [Google Scholar] [CrossRef]
- Celzard, A.; Mareche, J.F.; Payot, F.; Furdin, G. Electrical conductivity of carbonaceous powders. Carbon 2002, 40, 2801–2815. [Google Scholar] [CrossRef]
- Sanchez-Gonzalez, J.; Macias-Garcia, A.; Alexandre-Franco, M.F.; Gomez-Serrano, V. Electrical conductivity of carbon black under compressin. Carbon 2005, 43, 741–747. [Google Scholar] [CrossRef]
- Vargas, A.M.M.; Cazetta, A.L.; Garcia, C.A.; Moraes, J.C.G.; Nogami, E.M.; Lenzi, E.; Costa, W.F.; Almeida, V.C. Preparation and characterization of activated carbon from a new raw lignocellulosic material: Flamboyant (Delonix regia) pods. J. Environ. Manag. 2011, 92, 178–184. [Google Scholar] [CrossRef]
- Fuente, E.; Menendez, J.A.; Suarez, D.; Montes-Moran, M.A. Basic surface oxides on carbon materials: A global view. Langmuir 2003, 19, 3505–3511. [Google Scholar] [CrossRef]
- Schonner, J.; Buchheim, J.R.; Scholz, P.; Adelhelm, P. Boehm titration revisited (part I): Practical aspects for achieving a high precision in quantifyng oxygen-containing surface groups on carbon materials. C J. Carbon Res. 2018, 4, 21. [Google Scholar] [CrossRef] [Green Version]
- Takahata, T.; Toda, I.; Ono, H.; Ohshio, S.; Akasaka, H.; Himeno, S.; Kokubu, T.; Saitoh, H. Detailed structural analyses of KOH activated carbon from waste coffee beans. Jpn. J. Appl. Phys. 2009, 48, 117001. [Google Scholar] [CrossRef]
- Rodriguez Correa, C.; Kruse, A. Biobased functional carbon materials: Production, characterization, and applications—A review. Materials 2018, 11, 1568. [Google Scholar] [CrossRef] [Green Version]
- Yun, Y.S.; Park, M.H.; Hong, S.J.; Lee, M.E.; Park, Y.W.; Jin, H.J. Hierarchically porous carbon nanosheets from waste coffee grounds for supercapacitors. ACS Appl. Mater. Interfaces 2015, 7, 3684–3690. [Google Scholar] [CrossRef]
- Mohamed, M.G.; Atajde, E.C., Jr.; Matsagar, B.M.; Na, J.; Yamauchi, Y.; Wu, K.C.W.; Kuo, S.W. Construction hierarchically mesoporous/microporous materials based on block copolymer and covalent organic framework. J. Taiwan Inst. Chem. Eng. 2020, 112, 180–192. [Google Scholar] [CrossRef]
- Qian, M.; Xuan, X.Y.; Pan, L.K.; Gong, S.Q. Porous carbon electrodes from activated wastes coffee grounds for capacitive deionization. Ionics 2019, 25, 3443–3452. [Google Scholar] [CrossRef]
- Ramon-Goncalves, M.; Alcarez, L.; Perez-Ferreras, S.; Leon-Gonzalez, M.E.; Rosales-Conrado, N.; Lopez, F.A. Extraction of polyphenols and synthesis of new activated carbon from spent coffee grounds. Sci. Rep. 2019, 9, 17706. [Google Scholar] [CrossRef] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | C | H | N | S | “H:C” Ratio | “O:C” Ratio |
|---|---|---|---|---|---|---|
| Robusta (R) | 52.5 | 6.9 | 2.2 | <0.05 | 0.13 | 0.89 |
| Arabica (A) | 56.0 | 7.9 | 2.5 | <0.05 | 0.14 | 0.78 |
| Sample | Preliminary Washing | Activating Agent (Activator/Substrate Ratio w/w) | Pyrolysis Temperature (°C) | Yield (%) | Surface Area (m2/g) |
|---|---|---|---|---|---|
| RAC-1 1 | EtOH, 24 h RT + Hexane 24 h RT | KOH (1:1) | 800 | 15 | 629 |
| RAC-2 1 | EtOH, 24 h RT + Hexane 24 h RT | K2CO3 (1:1) | 600 | 13 | 765 |
| RAC-3 1 | H2O | none | 800 | 10 | 478 |
| RAC-4 1 | H2O | ZnCl2 (1:1) | 800 | 15 | 367 |
| AAC-1 | H2O | KOH (1:1) | 800 | 12 | 1407 |
| AAC-2 | H2O, 24 h RT + Hexane 24 h RT | KOH (1:1) | 800 | 12 | 1362 |
| AAC-3 2 | EtOH, 1 h RT | NaHCO3/CH3COOH (1:1:1) under stirring one night, RT; + KOH (1:1) | 800 | 12 | 1868 |
| AAC-4 2 | H2O, 24 h RT | KOH (1:1) | 800 | 10 | 1535 |
| AAC-5 | EtOH, 24 h RT + Hexane 24 h RT | KOH (1:1) | 600 | 15 | 615 |
| AAC-6 | EtOH, 24 h RT + Hexane 24 h RT | KOH (1:1) | 700 | 13 | 1715 |
| AAC-7 | EtOH, 24 h RT + Hexane 24 h RT | KOH (1:1) | 800 | 12 | 1375 |
| AAC-8 3 | EtOH, 24 h RT + Hexane 24 h RT | KOH (1:1) | 700 | 12 | 721 |
| AAC-9 4 | none | KOH (1:1) | 800 | 30 | 1200 |
| AAC-10 5 | EtOH, 24 h RT + Hexane 24 h RT | KOH (1:1) | 600 | 17 | 495 |
| M | Commercial: Merck, K39413913 002; activated charcoal, extra food pure grade | unknown | 823 | ||
| C | Commercial: CECA, Arkema, Acticarbone NCL 1240 | unknown | 932 | ||
| LP39 | Commercial: Jacobi®LP39® | unknown | 1782 | ||
| Sample | Density (g/cm3) | PZC |
|---|---|---|
| AAC-9 | 1.78 | 9.5 |
| M | 1.81 | 8.8 |
| C | 1.96 | 10.5 |
| LP39 | 1.44 | 6.7 |
| Sample | Size Lc 002 (Å) | Size Lc 100 (Å) | dspacing 002 (Å) | dspacing 100 (Å) |
|---|---|---|---|---|
| RAC-1 | 0.7 | 1.7 | 3.780 | 2.078 |
| RAC-2 | 0.8 | 1.7 | 3.795 | 2.060 |
| RAC-3 | 0.9 | 1.9 | 3.750 | 2.081 |
| RAC-4 | 1.2 | 1.8 | 3.760 | 2.062 |
| AAC-1 1 | - | 1.7 | - | 2.065 |
| AAC-2 | 1.3 | 2.0 | 3.740 | 2.076 |
| AAC-3 | 0.8 | 1.1 | 3.798 | 2.061 |
| AAC-4 | 1.1 | 1.8 | 3.839 | 2.071 |
| AAC-5 | 1.0 | 1.3 | 3.739 | 2.081 |
| AAC-6 | 0.9 | 1.5 | 3.716 | 2.081 |
| AAC-7 | 1.0 | 1.9 | 3.691 | 2.078 |
| AAC-8 | 0.8 | 1.6 | 3.792 | 2.074 |
| AAC-9 | 0.9 | 1.2 | 3.776 | 2.085 |
| AAC-10 | 0.8 | 1.4 | 3.749 | 2.088 |
| M | 0.8 | 2.0 | 3.536 | 2.068 |
| C | 1.1 | 2.2 | 3.759 | 2.073 |
| LP39 | 1.1 | 1.5 | 3.874 | 2.099 |
| Sample | C=C sp2 ~284.4 eV | C-C sp3 ~285.2 eV | C-OH ~285.8 eV | C=O ~286.7 eV | O-C=O ~288.1 eV | CO32− ~290.1 eV |
|---|---|---|---|---|---|---|
| RAC-1 | 57.5 | 9.2 | 9.3 | 8.2 | 5.3 | 10.6 |
| RAC-2 | 41.9 | 34.6 | 11.6 | 3.7 | 4.9 | 3.4 |
| RAC-4 | 37.4 | 32.6 | 7.1 | 12.7 | 6.2 | 4.0 |
| AAC-1 | 51.9 | 14.9 | 6.7 | 8.3 | 5.4 | 12.8 |
| AAC-2 | 48.0 | 26.3 | 11.6 | 8.6 | 5.5 | - |
| AAC-3 | 46.8 | 24.6 | 11.7 | 9.6 | 7.3 | - |
| AAC-4 | 56.4 | 7.1 | 11.2 | 8.7 | 5.9 | 10.7 |
| AAC-5 | 55.1 | 13.0 | 10.1 | 8.3 | 5.7 | 7.8 |
| AAC-6 | 54.4 | 12.0 | 9.4 | 10.3 | 5.6 | 8.3 |
| AAC-7 | 59.6 | 12.2 | 9.2 | 6.0 | 5.2 | 7.8 |
| AAC-8 * | 46.7 | 24.6 | 11.7 | 9,6 | 7.3 | - |
| AAC-9 | 37.3 | 36.8 | 9.5 | 5.6 | 3.9 | 6.9 |
| AAC-10 | 48.6 | 28.9 | 12.4 | 5.6 | 4.5 | - |
| M | 57.6 | 17.3 | 5.7 | 6.6 | 3.1 | 9.7 |
| C | 47.2 | 28.5 | 18.1 | 6.2 | - | - |
| LP39 | 42.8 | 34.7 | 8.5 | 4.6 | 3.8 | 5.7 |
| Sample | Csp3−OH 531.0 eV | C=O 532.2 eV | CAr−OH 533.5 eV |
|---|---|---|---|
| RAC-1 | 22.0 | 41.0 | 37.0 |
| RAC-2 | 33.8 | 43.0 | 23.2 |
| RAC-4 | 19.9 | 33.3 | 46.8 |
| AAC-1 | 12.7 | 51.1 | 36.2 |
| AAC-2 1 | 29.7 | 40.1 | 29.4 |
| AAC-3 1 | 22.1 | 40.9 | 28.4 |
| AAC-4 | 25.2 | 48.3 | 26.5 |
| AAC-5 | 22.9 | 52.2 | 24.8 |
| AAC-6 | 25.8 | 42.1 | 32.0 |
| AAC-7 | 33.6 | 44.7 | 21.7 |
| AAC-8 | 41.2 | 39.5 | 19.4 |
| AAC-9 | 35.9 | 49.8 | 14.3 |
| AAC-10 | 30.5 | 46.6 | 22.9 |
| M | 4,8 | 65,4 | 29,8 |
| C | 26.6 | 54.3 | 19.1 |
| LP39 | 34.5 | 47.1 | 18.4 |
| Sample | Carboxylic (mmol/g) | Lactonic (mmol/g) | Phenolic (mmol/g) | Acidic (mmol/g) | Basic (mmol/g) |
|---|---|---|---|---|---|
| AAC-5 | 1.71 | 0 | 0.80 | 2.34 | 2.51 |
| AAC-6 | 2.80 | 0 | 0.10 | 2.79 | 2.90 |
| AAC-7 | 2.77 | 0 | 0.03 | 2.80 | 3.20 |
| AAC-9 | 0.05 | 0 | 0.45 | 0.50 | 1.50 |
| M | 0 | 0 | 0.10 | 0.10 | 0.60 |
| C | 0 | 0 | 0 | 0 | 0.70 |
| LP39 | 0.49 | 0 | 0.51 | 1.00 | 0 |
| Sample | SBET (m2/g) | Vt (cm3/g) | Vm (cm3/g) | Vμ (cm3/g) | Vμ/Vt (%) | Dp (nm) |
|---|---|---|---|---|---|---|
| RAC-1 | 629 | 0.30 | 0.06 | 0.24 | 80.0 | 1.69 |
| RAC-2 | 765 | 0.32 | 0.05 | 0.27 | 84.8 | 1.64 |
| RAC-3 | 478 | 0.20 | 0.03 | 0.17 | 85.0 | 1.69 |
| RAC-4 | 367 | 0.15 | 0.02 | 0.13 | 86.7 | 1.61 |
| AAC-1 | 1407 | 0.76 | 0.08 | 0.68 | 89.5 | 2.16 |
| AAC-2 | 1362 | 0.72 | 0.11 | 0.61 | 84.7 | 2.13 |
| AAC-3 | 1868 | 1.06 | 0.44 | 0.62 | 58.5 | 2.27 |
| AAC-4 | 1535 | 0.82 | 0.12 | 0.70 | 85.4 | 2.14 |
| AAC-5 | 615 | 0.32 | 0.01 | 0.31 | 96.9 | 2.10 |
| AAC-6 | 1715 | 0.92 | 0.07 | 0.85 | 92.4 | 2.14 |
| AAC-7 | 1375 | 0.74 | 0.07 | 0.67 | 90.5 | 2.14 |
| AAC-8 | 721 | 0.31 | 0.01 | 0.30 | 96.8 | 2.12 |
| AAC-9 | 1200 | 0.50 | 0.07 | 0.43 | 86.0 | 1.67 |
| AAC-10 | 495 | 0.27 | 0.01 | 0.26 | 96.3 | 2.18 |
| M | 823 | 0.52 | 0.14 | 0.38 | 73.1 | 2.54 |
| C | 932 | 0.49 | 0.04 | 0.45 | 91.9 | 2.11 |
| LP39 | 1782 | 1.62 | 1.14 | 0.51 | 31.5 | 3.64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosson, E.; Sgarbossa, P.; Mozzon, M.; Venturino, F.; Bogialli, S.; Glisenti, A.; Talon, A.; Moretti, E.; Carturan, S.M.; Tamburini, S.; et al. Novel Correlations between Spectroscopic and Morphological Properties of Activated Carbons from Waste Coffee Grounds. Processes 2021, 9, 1637. https://doi.org/10.3390/pr9091637
Rosson E, Sgarbossa P, Mozzon M, Venturino F, Bogialli S, Glisenti A, Talon A, Moretti E, Carturan SM, Tamburini S, et al. Novel Correlations between Spectroscopic and Morphological Properties of Activated Carbons from Waste Coffee Grounds. Processes. 2021; 9(9):1637. https://doi.org/10.3390/pr9091637
Chicago/Turabian StyleRosson, Egle, Paolo Sgarbossa, Mirto Mozzon, Federico Venturino, Sara Bogialli, Antonella Glisenti, Aldo Talon, Elisa Moretti, Sara Maria Carturan, Sergio Tamburini, and et al. 2021. "Novel Correlations between Spectroscopic and Morphological Properties of Activated Carbons from Waste Coffee Grounds" Processes 9, no. 9: 1637. https://doi.org/10.3390/pr9091637
APA StyleRosson, E., Sgarbossa, P., Mozzon, M., Venturino, F., Bogialli, S., Glisenti, A., Talon, A., Moretti, E., Carturan, S. M., Tamburini, S., Famengo, A., da Costa Ribeiro, A. P., Benhabiles, S., Kamel, R., Zorzi, F., & Bertani, R. (2021). Novel Correlations between Spectroscopic and Morphological Properties of Activated Carbons from Waste Coffee Grounds. Processes, 9(9), 1637. https://doi.org/10.3390/pr9091637