
Abstract
Although the fact that genetic predisposition and environmental exposures interact to shape development and function of the human brain and, ultimately, the risk of psychiatric disorders has drawn wide interest, the corresponding molecular mechanisms have not yet been elucidated. We found that a functional polymorphism altering chromatin interaction between the transcription start site and long-range enhancers in the FK506 binding protein 5 (FKBP5) gene, an important regulator of the stress hormone system, increased the risk of developing stress-related psychiatric disorders in adulthood by allele-specific, childhood trauma–dependent DNA demethylation in functional glucocorticoid response elements of FKBP5. This demethylation was linked to increased stress-dependent gene transcription followed by a long-term dysregulation of the stress hormone system and a global effect on the function of immune cells and brain areas associated with stress regulation. This identification of molecular mechanisms of genotype-directed long-term environmental reactivity will be useful for designing more effective treatment strategies for stress-related disorders.
Similar content being viewed by others

Main
Epidemiological, family and molecular genetic studies have shown that genetic predisposition as well as stressful or traumatic life events, especially in childhood, are important risk factors for psychiatric disorders, including major depression and post-traumatic stress disorder (PTSD) and that these factors most likely have interactive, rather than additive, effects1. Although specific gene × environment interactions have been described2,3,4,5, the molecular basis of gene × environment interaction in mood and anxiety disorders remains obscure. FKBP5 is an important functional regulator of the glucocorticoid receptor complex6. The glucocorticoid receptor is a pivotal nuclear receptor of the stress hormone system mediating the negative feedback of this axis to terminate the stress response after the end of a threat7. Dysregulation in this system has been described in stress-related psychiatric disorders and as a long-term consequence of exposure to early life trauma8,9. FKBP5 alters glucocorticoid receptor function by decreasing ligand binding and impeding translocation of the receptor complex to the nucleus10,11. Furthermore, FKBP5 is part of an intracellular ultra-short negative feedback loop that regulates glucocorticoid receptor activity. Glucocorticoid receptor activation induces FKBP5 transcription via activation at predominantly intronic steroid hormone response elements12, leading to increased transcription of FKBP5, entailing restrained glucocorticoid receptor activity. We and others have shown that polymorphisms in FKBP5 (haplotypes including rs1360780, rs9296158, rs3800373 and rs9470080) interact with early trauma or childhood abuse to predict adult PTSD, suicide attempts and major depression3,13,14,15,16,17. Here we identified a molecular mechanism for this gene × environment interaction by long-term epigenetic modifications.
Results
rs1360780 moderates the risk for PTSD after early trauma
Previously, we found that the same FKBP5 polymorphisms that interact with early trauma are also associated with altered induction of FKBP5 mRNA by glucocorticoid receptor stimulation in peripheral blood18. We hypothesized that the associated functional variant lies in or close to glucocorticoid response elements (GREs) in the FKBP5 locus. Given that the originally genotyped variants are in high linkage disequilibrium over the entire locus, we used genotype data from Illumina OmniExpress Single Nucleotide Polymorphism (SNP) arrays spanning the whole FKBP5 locus in 192 individuals of the Grady trauma project and imputed all currently known variants using the 1,000 Genomes project data (http://www.1000genomes.org/)19. This resulted in 799 polymorphisms imputed with a quality greater than 0.6 and an average quality score of 0.93. Linkage disequilibrium with rs1360780, rs9296158, rs9470080 and rs3800373 was evaluated using the tagger software implemented in Haploview version 4.2. Of the imputed polymorphisms, 48 had an r2 greater than 0.4 with at least one of the four polymorphisms mentioned above to interact with early trauma and these variants spanned 192 kb of the locus (Supplementary Table 1). Of all genotyped and imputed variants tagging this association, rs1360780, the variant originally identified as being associated with differential induction of FKBP5 by glucocorticoid receptor activation18, was the SNP in the tagging bin that was located closest to a functional GRE (see detailed description below).
In an expanded African-American cohort (N = 1,963) from the Grady trauma project (which includes 1,194 more individuals than were present in our original report3), we first confirmed and extended our previously reported interaction of FKBP5 rs1360780 and early trauma on adult PTSD symptoms. We not only observed a significant interaction of child abuse, determined by the Childhood Trauma Questionnaire (CTQ) and FKBP5 rs1360780, on current adult PTSD symptoms, as measured by the modified PTSD symptom scale (mPSS total score, F1963,2 = 4.40, P = 0.012), but also on lifetime PTSD, as assessed by the criterion standard diagnostic instrument, the clinician-administered PTSD scale (CAPS, N = 519). In a logistic regression analysis, both early trauma (P < 0.001) and the interaction of early trauma and the FKBP5 rs1360780 risk allele carrier status (P = 0.034) were significant predictors for lifetime PTSD without main genetic effect of FKBP5. The risk of suffering from lifetime PTSD was significantly increased by exposure to early trauma in FKBP5 risk allele carriers (χ2 = 28.6, degrees of freedom = 2, P < 0.001), but not in carriers of the protective genotype (χ2 = 2.02, degrees of freedom = 2, P = 0.36) (Fig. 1). These data lend further support to the robustness of the moderation of child abuse–related risk for adult PTSD by rs1360780.

Shown is the interaction of child abuse and FKBP5 rs1360780 protective genotype (left) or risk allele carrier status (right) on percentage lifetime PTSD (CAPS). FKBP5 protective genotype: no abuse, N = 133; one type, N = 27; two types, N = 16; FKBP5 risk allele carriers: no abuse; N = 252, one type, N = 69; two types, N = 22 (n.s. P > 0.05, ***P < 0.001).
rs1360780 affects FKBP5 chromatin shape and transcription
The glucocorticoid receptor transcriptionally regulates FKBP5 mainly by distal intronic GREs20. rs1360780 is located in a functional enhancer region and 488 bp from a GRE in intron 2. To test whether this SNP itself has the potential to alter FKBP5 transcription, we cloned reporter gene constructs encompassing the consensus GRE site together with either the rs1360780 A/T risk allele or the C/G protective allele (Fig. 2a). The construct with the risk allele exhibited stronger activity than the construct with the protective allele, both in the absence of glucocorticoid receptor (1.4-fold, P < 0.001) and in the presence of activated glucocorticoid receptor (1.7-fold, P = 0.036) (Fig. 2b,c). These data are consistent with previously reported in vivo data showing a stronger correlation of FKBP5 mRNA and plasma cortisol in peripheral blood cells from risk genotype carriers as compared with carriers of the protective allele18. In fact, the sequence containing the A/T risk allele of rs1360780 is predicted to form a putative TATA box, possibly enhancing gene transcription as compared to the C/G allele. The A allele indeed exhibited a stronger TATA box binding protein (TBP) binding than the G allele (P = 0.009; Fig. 2d). Data from chromatin immunoprecipitation experiments suggest that this distal GRE comes into contact with RNA polymerase and, likely, the transcription start site (TSS), supporting a functional consequence of altered TBP binding in intron 2 on FKBP5 mRNA transcription20. Taking advantage of the chromatin conformation capture technique, we found an rs1360780 genotype–independent interaction of intron 7 with the TSS via three-dimensional loop formation (Fig. 3 and Supplementary Fig. 1a–d) and a genotype-dependent interaction of intron 2 and the TSS in a homozygote risk (AA) allele carrier, but not a carrier of the protective (GG) genotype. These results support the theory that the functional effects reported for the FKBP5 haplotype on FKBP5 mRNA and protein levels could be mediated by the different chromatin conformation and, thus, transcriptional effects of the two opposite alleles of rs1360780 (ref. 18).

(a) Representation of the reporter construct. The luciferase gene is driven by a basal human elongation factor-1 alpha (EF-1α, EEF1A1) promoter with a 802-bp fragment of FKBP5 intron 2 containing the putative GRE and rs1360780 cloned in front of the promoter. The rs1360780 risk allele A/T might form a TATA box–like sequence, which would enhance transcriptional activity. (b) Allele specificity of the reporter gene activity in glucocorticoid receptor–free HEK 293 cells. The reporter activity was reduced for the protective allele versus the risk allele (***P < 0.001, Student's t test, unpaired, two sided). (c) Stimulation with 50 nM dexamethasone significantly enhanced intron 2–driven luciferase activity in glucocorticoid receptor–expressing HeLa cells in a genotype-dependent manner. Reduced activity was observed for the protective allele versus the risk allele (*P = 0.036, Student's t test, unpaired, two sided). Baseline luciferase activity in HeLa cells without dexamethasone stimulation did not reveal significant differences (data not shown). In the box plots, the box extends indicate lower quartile and upper quartile, and the whiskers denote sample minimum and maximum. The line in the box represents the median and dots represent outliers. (d) Relative binding of recombinant TBP to a double-stranded 60-mer oligonucleotide containing either the A or G allele of rs1360780. The A allele exhibited a stronger binding to TBP than the G allele. (**P = 0.009, Student's t test, unpaired, two sided). The assay was run in triplicates, background activity was subtracted from the measurement reading. Data are expressed as mean ± s.e.m.

(a) Top, FKBP5 genomic locus with the TSS indicated by an arrow and intron 2 and intron 7 indicated by black boxes. The upright thin lines represent EcoRI cutting sites of genomic DNA. Bottom, chromatin conformation capture interaction data. Amplicon primers are indicated by small arrows, and the fragments containing the TSS, intron 2 and intron 7 are emphasized by gray boxes. Data from quantitative PCR are plotted as relative crosslinking frequency on the y axis. A specific interaction was detected as the local peak in interaction frequencies. Thus, intron 7 showed a stronger interaction with the TSS than with fragments upstream and downstream of the intron 7 fragment in both cell lines. Moreover, we observed a strong local peak for the interaction of the TSS with intron 2, but only in the cell line carrying the risk (AA) genotype. This confirmed the physical interaction of the TSS with intron 7 and intron 2 in a genotype-dependent manner. (b,c) Three-dimensional interaction of intronic GREs in intron 2 and 7 of FKBP5 with the TSS. FKBP5 mRNA transcription is induced by cortisol via a three-dimensional interaction and loop formation of predominantly distal enhancer regions (blue) harboring GREs with the core promoter site (PolII = RNA polymerase II). The interaction of intron 2 with the TSS in risk allele carriers leads to an increased FKBP5 induction in response to glucocorticoid receptor activation (represented by a red arrow).
Consistent with the ultra-short negative feedback between FKBP5 and glucocorticoid receptor activation, this genotype-mediated increased FKBP5 transcription has been shown to be associated with glucocorticoid receptor resistance in healthy controls3,15 and with a decreased efficiency of negative feedback of the stress hormone axis accompanied by prolonged cortisol release21. We sought to unravel the molecular mechanism by which a genetic predisposition to react more strongly to a stressor in the short term interacts with exposure to childhood trauma to increase the risk of developing psychiatric disorders in the long term.
DNA demethylation in FKBP5 in traumatized individuals
Epigenetic changes, especially changes in DNA methylation, have been reported as long-lasting consequences of early trauma22,23,24, and glucocorticoid receptor activation has been shown to induce local changes in DNA methylation at GREs, including in the murine Fkbp5 locus25,26. We hypothesized that excessive cortisol release following early life stress exposure in FKBP5 risk allele carriers would lead to epigenetic changes in the GREs of FKBP5, resulting in lasting disruptions of the ultra-short feedback loop that balances FKBP5 and glucocorticoid receptor activity, entailing dysregulation of the stress hormone system, and ultimately increasing the risk for certain psychiatric disorders.
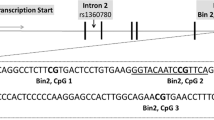
To investigate the possibility that FKBP5 × early trauma interactions are mediated by epigenetic modifications, we performed DNA methylation analysis by pyrosequencing of bisulfite-treated genomic DNA extracted from peripheral blood cells from individuals selected from the Grady trauma project. DNA from individuals having experienced both sexual and physical child abuse were compared with DNA from individuals without any childhood trauma (N = 76; high trauma cohort, N = 30; control cohort, N = 46; Table 1). We screened the CpG island in the vicinity of the TSS, as well as the regions around functional GREs20 in the putative promoter region and introns 2, 5 and 7 for the extent of CpG methylation (Fig. 4a and Supplementary Table 2). We observed significant DNA methylation higher than 5% in regions close to the GREs in the promoter region (30–100%), intron 2 (30–93%) and intron 7 (29–100%), but not in the CpG island or the two GREs in intron 5 (<5%). In intron 7, but not in the two other regions, two methylated CpGs were located in consensus GRE sequences.

(a) Significant DNA methylation was observed in the promoter region, intron 2 and intron 7 of FKBP5, as indicated in green. (b) Single CpG site methylation in the four groups (early trauma × FKBP5 rs1360780 carriers). Data are expressed as mean ± s.e.m.
We next tested the effects of FKBP5 genotype (rs1360780 risk allele carrier model) and exposure to childhood abuse (CTQ categorical scale), as well as their interaction, on the extent of DNA methylation in these three regions, summarizing the percentage methylation of these 16 CpGs into seven bins of one to three CpGs to a mean methylation score according to their spatial proximity to the consensus GRE sites (Fig. 4b and Supplementary Fig. 2). The analyses were corrected for age and gender as covariates and multiple testing. In one bin of intron 7 (bin 2), but not the other bins, significant early trauma (F73,1 = 8.2, P = 0.006), genotype (F73,1 = 34.33, P < 0.001) and interaction effects between genotype and early trauma (F73,1 = 31.01, Pcorr < 0.001) on methylation were observed. This bin contains three CpGs, with one CpG located in the consensus GRE in intron 7 (Supplementary Fig. 2). An average decrease of 12.3% in DNA methylation in these three CpGs was detected in child abuse–exposed risk allele carriers compared to the other three groups (Supplementary Fig. 3). When correlating levels of child abuse measured using the log-transformed total CTQ score and the subscores for physical, emotional and sexual abuse on this scale, with the mean methylation of this CpG bin in intron 7, we found significant differences in the correlation coefficients between the risk allele carriers (N = 55) and the protective genotype carriers (N = 19) (R = −0.646, P < 0.001 for risk allele and R = 0.414, P = 0.078 for protective genotype carriers and total CTQ with a Fisher z score of –4.23 and P = 7.0 × 10−5; Fig. 5 and Supplementary Table 3). Heterozygous individuals carrying one risk allele did not differ from homozygous risk allele carriers (Supplementary Fig. 4). This emphasizes the effects of early trauma severity on FKBP5 demethylation in risk allele carriers, but not in protective genotype carriers.

Correlation between intron 7 bin 2, mean methylation and log-transformed CTQ scores by FKBP5 rs1360780 genotype in the Grady and Conte cohort are shown. (a) Grady cohort. Risk allele carriers exhibited a strong negative correlation (R = −0.646, P < 0.001) between methylation and CTQ total load compared with carriers of the protective genotype (R = 0.414, P = 0.078) (Fisher z score = −4.23, P < 0.001). (b) Conte cohort. Correlation between methylation and total CTQ in risk allele carriers (R = −0.273, P = 0.124), and in carriers of the protective genotype (R = 0.153, P = 0.485) (Fisher z score = −1.5, P = 0.133). (c) Grady cohort. Negative correlation was found between methylation and the CTQ physical abuse subscore in risk allele carriers (R = −0.586, P < 0.001), but not in carriers of the protective genotype (R = 0.360, P = 0.130) (Fisher z score = −4.49, P < 0.001). (d) Conte cohort. Negative correlation was observed between methylation and the CTQ physical abuse subscore in risk allele carriers (R = −0.397, P = 0.022), but not in carriers of the protective genotype (R = 0.246, P = 0.258) (Fisher z score = −2.33, P = 0.019). (e) Grady cohort. Negative correlation was found between methylation and the CTQ emotional abuse subscore in risk allele carriers (R = −0.685, P < 0.001), but not in carriers of the protective genotype (R = 0.321, P = 0.181) (Fisher z score = −4.1, P < 0.001). (f) Conte cohort. Negative correlation was found between methylation and the CTQ emotional abuse subscore in risk allele carriers (R = −0.397, P = 0.022), but not in carriers of the protective genotype (R = 0.022, P = 0.922) (Fisher z score = −1.53, P = 0.126). (g) Grady cohort. Negative correlation was found between methylation and the CTQ sexual abuse subscore in risk allele carriers (R = −0.656, P < 0.001), but not in carriers of the protective genotype (R = 0.599, P = 0.007) (Fisher z score = −5.17, P < 0.001). (h) Conte cohort. Negative correlation was found between methylation and the CTQ sexual abuse subscore in risk allele carriers (R = 0.118, P = 0.514), and in carriers of the protective genotype (R = 0.305, P = 0.922) (Fisher z score = −0.68, P = 0.496).
To test whether the observed effects of child abuse may have been confounded by the increased exposure to more recent types of trauma in the abused group (Table 1), we explored the correlation of intron 7 bin 2 DNA methylation and the number of exposures to adult trauma separately in risk alleles carriers with or without child abuse. We did not observe a correlation of exposure to adult trauma with intron 7 bin 2 DNA methylation in either the childhood abuse risk allele group (R = 0.020, P = 0.933, N = 20) or the control risk allele group (R = 0.018, P = 0.917, N = 35). This suggests that the observed effect of early trauma on DNA methylation is independent of subsequent exposure to trauma (Supplementary Fig. 5).
We next investigated the relationship of severity of early trauma and FKBP5 DNA methylation in a second female-only cohort recruited as part of a separate study investigating the effects of early trauma on biological markers4,27 (Table 1). In this cohort, we correlated the log-transformed total CTQ score as well as the subscores for physical, emotional and sexual abuse with the mean DNA methylation of the three CpGs in the second bin of intron 7. In FKBP5 risk genotype carriers (N = 33), we observed a negative correlation between the methylation score and the total CTQ score (R = −0.273, P = 0.124), the physical abuse subscore (R = −0.397, P = 0.022) and the emotional abuse subscore (R = −0.397, P = 0.022), but not the subscore for sexual abuse (R = 0.118, P = 0.514) (Fig. 5). In carriers of the protective genotype (N = 23), these correlations were either absent or positive (Supplementary Table 3). The difference in correlations was significant for the physical abuse subscore (Fischer z score = −2.33, P = 0.019). These data from an independent sample support the notion of allele-specific demethylation of CpGs close to and in functional GREs in intron 7 of the FKBP5 gene with early trauma exposure. The difference in effect size and relative contributions of different trauma types is likely a result of the fact that this second cohort was exposed to less overall trauma, including childhood abuse, than the cohort from the Grady trauma project (Table 1). In addition, the replication cohort was ethnically diverse, but the effects of childhood abuse on DNA methylation were consistent across the two main ethnic groups, European and African Americans.
To rule out the possibility that trauma- or psychiatric disease–related differences in immune cell fractions confound the reported results28, we reran the analysis in 41 women of the replication cohort (21 risk allele carriers, 20 protective genotype carriers) in whom we also had data on the relative amount of the main immune cell subtypes (CD14-positive monocytes and neutrophils, CD4- and CD8-positive lymphocytes, and CD16- and CD56-positive NK cells). Using these as covariates, we found that the differences in correlations between CTQ scores and intron 7 methylation in the two genotype groups remained substantial. For physical abuse, the corrected correlations were R = −0.476 in risk allele carriers and R = 0.40 in carriers of the protective genotype, with a significant z score of 2.78 (P = 0.005). In addition, we did not observe any correlations between the relative amount of immune cell subtypes and intron 7 DNA methylation, suggesting that possible trauma-related differences in these cell types are not likely to confound our results. However, we cannot exclude the effect of immune cell subtypes that were not specifically tested.
Demethylation of FKBP5 in neuronal cells
Our data therefore point to an allele-specific, early trauma exposure–dependent demethylation of CpGs close to and in GREs in intron 7 of FKBP5 (Supplementary Fig. 1a–d). To determine whether this demethylation could be related to excessive glucocorticoid receptor activation and might also be seen in tissues other than peripheral blood, including neuronal tissue, we used a multipotent human hippocampal progenitor cell line in which exposure to dexamethasone has been shown to be accompanied by a reduction in cell proliferation and neuronal differentiation29. Cells were exposed to dexamethasone during both the proliferation and differentiation phases, similar to previous studies29. The percentage of DNA methylation across CpGs in introns 2 and 7 in these neuronal progenitor cells was comparable to that in peripheral blood cells. Exposure to dexamethasone led to a highly significant (P < 0.001) DNA demethylation in CpGs in intron 7, but not intron 2 (Fig. 6a). In fact, the CpG bin in intron 7 that was affected by early trauma in FKBP5 risk allele carriers showed the strongest DNA demethylation following glucocorticoid receptor stimulation in hippocampal progenitor cells (average of 17.1% demethylation in these three CpGs, P < 0.001 for differentiation phase, which includes proliferation). The observed demethylation in intron 7 remained unchanged after 20 days in culture in steroid-devoid medium, supporting a stable epigenetic memory of glucocorticoid-induced demethylation (Fig. 6b). Moreover, glucocorticoid receptor activation by dexamethasone treatment after proliferation and differentiation did not result in demethylation in intron 7 bin 2, suggesting a sensitive period of time for glucocorticoids-mediated epigenetic changes (Fig. 6c).

(a) Dexamethasone treatment induced demethylation in the human hippocampal progenitor cell line HPC03A/07 in the proliferation and differentiation phase in intron 7 (***P < 0.001), but not intron 2. Data are expressed as mean ± s.e.m. (b) Dexamethasone treatment of human hippocampal progenitor cells in proliferation and differentiation phase resulted in significant demethylation of CpGs in FKBP5 intron 7, bins 2 and 3 (***P < 0.001, **P = 0.01, n.s. P > 0.05, Student's t test, unpaired, two-sided). Dexamethasone treatment in the proliferation phase only resulted in similar demethylation of CpGs in FKBP5 intron 7 in comparison with the differentiation phase (data not shown). A subsequent washout of dexamethasone and an additional 20-d incubation revealed similar results, supporting a long-lasting demethylation in bins 2 and 3. Data are expressed as mean ± s.e.m. (c) Treatment in proliferation and differentiation phase resulted in an immediate demethylation of intron 7 bin 2 (***P < 0.001, Student's t test, unpaired, two-sided). Treatment in the proliferation and differentiation phase and subsequent washout phase in steriod-devoid medium for 20 d revealed a stable demethylation by glucocorticoid receptor activation. In contrast, dexamethasone treatment after proliferation and differentiation did not induce a long-lasting demethylation in intron 7 bin 2 (n.s. P = 0.063, Student's t test, unpaired, two-sided), suggesting a sensitive period of time for glucocorticoids-mediated epigenetic changes. Data are expressed as mean ± s.e.m.
The existence of a sensitive time period for these epigenetic effects was also apparent from data from human blood cells, as only exposure to child abuse, but not later trauma, predicted FKBP5 demethylation (see above), and there was a lack of correlation of serum cortisol levels and DNA methylation in blood samples from both human cohorts taken at the same time point (Grady trauma project, R = 0.083, P = 0.489, N = 72; Conte replication cohort, R = 0.069, P = 0.603, N = 59; Supplementary Fig. 6). These data further substantiate the conclusion that these specific CpGs in intron 7 are sensitive to stable, glucocorticoid receptor–mediated demethylation during a vulnerable time period and that our findings of early trauma–and genotype-dependent demethylation also pertain to tissues beyond peripheral blood.
Functional effects of intron 7 demethylation
We then investigated whether the observed changes in DNA methylation in intron 7 alter the glucocorticoid responsiveness of FKBP5 in vitro and in vivo. We inserted 515 bp of intron 7, encompassing the three functional GREs, into a CpG-free reporter plasmid30 (Fig. 7a) and assessed its activity in HeLa cells as a function of insert methylation. No effect of methylation was observed in the absence of glucocorticoid receptor activation, which is consistent with the absence of correlation between intron 7 methylation and baseline FKBP5 expression in peripheral blood cells (data not shown). However, methylation significantly attenuated the response to 50 nM dexamethasone (relative induction = 19-fold in unmethylated versus 13-fold in methylated construct, P = 0.035; Fig. 7b). These data indicate that methylation of CpGs surrounding the functional GREs in intron 7 decreases the induction of FKBP5 by glucocorticoid receptor activation without affecting baseline activity.

(a) Representation of the reporter construct. A 514-bp fragment of FKBP5 intron 7 containing three putative GREs was cloned in front of the EF-1α promoter and the luciferase gene, acting as transcriptional regulator. Only the FKBP5 insert can be methylated in vitro by MSssI, as the vector is CpG free. Circled M denotes putative methylation sites. Two of them are located in consensus GRE sequences. (b) Stimulation by 50 nM dexamethasone was attenuated by DNA methylation (*P = 0.035, n.s. P = 0.146, Student's t test, unpaired, two-sided). Baseline luciferase activity without dexamethasone stimulation did not reveal significant differences (P > 0.05; data not shown). In the box plots, the box extends indicate lower quartile and upper quartile, and the whiskers denote sample minimum and maximum. The line in the box represents the median and dots represent outliers. (c) Methylation in intron 7 was correlated with dexamethasone-mediated inhibition of lipopolysaccharide-induced interleukin 6 production (DEX IC50) in peripheral blood monocytes ex vivo (R = −0.531, P = 0.002). This correlation was stronger in rs1360780 risk allele carriers (N = 19, R = −0.691, P = 0.001) than carriers of the protective genotype (N = 13, R = −0.119, P = 0.698). This suggests that lower DNA methylation in intron 7 leads to stronger FKBP5 induction and to glucocorticoid receptor resistance. (d) Exposure to early trauma enhanced FKBP5 genotype–dependent differences in glucocorticoid receptor–dependent gene expression in peripheral blood. Top, R scores of the correlation of peripheral blood gene expression and cortisol for the 76 transcripts with significant genotype-dependent correlation differences in individuals with high levels of early trauma (cases, N = 55) and stratified by FKBP5 genotype (risk allele: N = 40, CTQ total score = 67.88 ± 14.71; protective genotype: N = 15, CTQ total = 72.44 ± 17.68). For all transcripts, the correlation with cortisol, and therefore the presumable responsiveness to the glucocorticoid receptor, was higher in FKBP5 protective genotype carriers (black squares) with a mean R of 0.74 than in carriers of the risk allele (open squares), with a mean R of 0.23. Bottom, correlation between gene expression levels and cortisol of the same transcripts, but in individuals that were not exposed to early trauma (controls, N = 74; risk allele: N = 60, CTQ total = 28.42 ± 2.96; protective genotype: N = 14, CTQ total = 28.79 ± 3.21). In this group, no genotype-dependent differences of the correlation were observed, with mean R scores of 0.31 and 0.35 for risk and protective genotypes, respectively. x axis represents individual transcripts 1 to 76.
To test whether intron 7 DNA methylation alters the ultra-short feedback loop between glucocorticoid receptor and FKBP5, and thereby alters glucocorticoid receptor sensitivity, we correlated the mean methylation score of intron 7 bin 2 with glucocorticoid receptor sensitivity measured ex vivo in peripheral blood monocytes in 32 individuals from the replication cohort. We observed a strong negative correlation between the half-maximal inhibitory concentration (IC50) for dexamethasone-mediated inhibition of lipopolysaccharide-induced interleukin 6 production in these cells (N = 32, R = −0.531, P = 0.002), which is a measure of glucocorticoid receptor sensitivity. This suggests that, as expected, less methylation of intron 7 CpGs is associated with higher induction of FKBP5 by glucocorticoid receptor activation, especially in risk allele carriers, representing an enhancement of the ultra-short feedback loop leading to increased glucocorticoid receptor resistance (Fig. 7c).
Global effect on gene transcription and brain structure
FKBP5 risk allele carrier status and early trauma exposure lead to demethylation of intron 7 CpGs in FKBP5, which further amplifies genotype-dependent differences in the FKBP5 and glucocorticoid receptor ultra-short feedback loop and, thus, glucocorticoid receptor sensitivity. FKBP5 genotype–dependent differences in the global regulation of glucocorticoid receptor–sensitive genes should therefore be amplified in trauma-exposed risk allele carriers compared with protective genotype carriers. To test this hypothesis, we examined the effects of FKBP5 rs1360780 genotype × environment interaction on peripheral blood mRNA expression of glucocorticoid receptor–responsive genes, as measured by gene expression arrays, in 129 individuals (child abuse + risk allele, N = 40; child abuse + protective genotype, N = 15; no child abuse + risk allele, N = 60; no child abuse + protective genotype, N = 14)15. In all 129 individuals, 1,627 transcripts showed a significant correlation (P < 0.05) with plasma cortisol concentrations, suggesting that they are responsive to glucocorticoid receptor activation. We found significant differences in the correlation of 76 of these transcripts with cortisol plasma levels when stratifying by FKBP5 genotype in individuals with child abuse (Fisher z score ≥ 1.96; Supplementary Table 4). For these 76 transcripts, the mean absolute correlation coefficient with plasma cortisol was R = 0.23 in the risk allele carriers with child abuse (that is, those exhibiting a demethylation of FKBP5 intron 7) as compared with R = 0.74 in the carriers of the protective genotype with child abuse (in which intron 7 methylation remained largely stable). This indicates a relative glucocorticoid receptor resistance in the trauma-exposed FKBP5 risk allele versus protective genotype carriers. These 76 transcripts did not show a genotype-dependent difference in correlation coefficients in individuals that were not exposed to child abuse (Fig. 7d), suggesting that exposure to early trauma enhances the FKBP5 genotype–dependent effect of glucocorticoid receptor sensitivity, most likely by epigenetic mechanisms. The genes most strongly affected by FKBP5 genotype for their correlation with plasma cortisol levels in trauma-exposed individuals showed a significant over-representation (WikiPathways, P < 0.01) of transcripts in the T cell receptor signaling pathway (Padjusted = 5.15 × 10−7), the TGF-β signaling pathway (Padjusted = 0.0007), the Wnt signaling pathway and pluripotency (Padjusted = 0.0044), and the inflammatory response pathway (Padjusted = 0.0099)31. These findings suggest that the combination of FKBP5 risk allele carrier status and early trauma exposure alters the stress hormone–dependent regulation of several genes in peripheral blood cells, and might thereby enhance the reported association of early trauma with immune and inflammatory dysregulation, further promoting system-wide symptoms of stress-related disorders32,33.
Structural imaging data from a subset of the replication cohort revealed that peripheral blood FKBP5 methylation significantly correlated with the volume of the right hippocampal head (N = 34, R = −0.484, Pcorr = 0.014; Supplementary Fig. 7). Despite the smaller N values for the structural imaging data in the less-traumatized replication cohort, these preliminary data suggest that FKBP5 demethylation is not only associated with an altered sensitivity of peripheral glucocorticoid receptors and more global changes in immune cell gene expression, but also with morphological changes in the brain indicative of a higher stress hormone system reactivity, thereby bridging the gap between abuse-related peripheral methylation changes and effects in the CNS.
Discussion
Here we provide evidence for an epigenetic mechanism that mediates the combined effects of environmental exposure in early life and a genetic polymorphism on the risk of developing stress-related psychiatric disorders. According to this mechanism, genetic variations shaped by evolution can determine the environmental adaption during the lifetime of an individual via epigenetic processes. Our data suggest the following scenario for an FKBP5 × child abuse interaction (Supplementary Fig. 1a–d): genetic differences lead to divergent chromatin conformations and interactions of long-range enhancers with the TSS, resulting in a differential transcriptional activation of FKBP5 by glucocorticoid receptor activation in response to childhood abuse. These changes in chromatin structure include regions around distal GREs and, together with increased cortisol levels and thus glucocorticoid receptor binding, lead to changes in DNA methylation in intron 7, further increasing the differential responsiveness of FKBP5 to glucocorticoid receptor activation. When installed during developmentally critical periods, these epigenetic changes remain stable over time. This model describes how epigenetic mechanisms stabilize and, in an environment-dependent manner, further amplify differential activities that were originally bestowed on the enhancer complex by genetic differences.
On a systems level, these molecular changes further tighten the ultra-short feedback loop between FKBP5 and the glucocorticoid receptor leading to glucocorticoid receptor resistance. Enhanced FKBP5 responsiveness would not only lead to long-term changes in stress hormone system regulation, but also to alterations of neuronal circuits, as reflected by structural changes in the hippocampus, and other glucocorticoid receptor responsive systems, as reflected by global gene expression changes in the immune system, resulting in a higher risk for trauma-associated psychiatric, immune and metabolic disorders in exposed adults. The fact that the interaction of FKBP5 genotype and early trauma can predispose an individual to both PTSD and depression indicates that this interaction is likely associated with stress sensitivity in general, crossing current diagnostic borders. Our findings appear to be of particular relevance for the developing organism, as the effects on DNA methylation seemed to be restricted to exposure to childhood trauma and were not influenced by traumatic experiences in adulthood or current cortisol levels, suggesting that there is a sensitive period in early development for these epigenetic effects. The importance of disinhibition of FKBP5 for mood and anxiety disorders is supported by rodent models showing that enhanced transcription of Fkbp5 in the amygdala is required for stress-, neuropsin- and EphB2-dependent induction of anxiety-like behavior in mice34. In addition, stress responsiveness is impaired in Fkbp5 knockout animal35,36.
In our studies, we focused on adverse environmental exposures. It is, however, possible that the described polymorphisms define not only risk versus resilience, but possibly environmentally reactive versus less reactive individuals. This would imply that the so-called risk-allele carriers may also profit more from positive environmental change. Our results provide insight into the molecular mechanisms and consequences of this gene × environment interaction, facilitating a better understanding of the pathophysiology of stress-related psychiatric disorders by including genetic and environmental information in the diagnosis, and potentially aiding in the development of new treatments targeting this mechanism.
Methods
Human samples.
The initial patient cohort that we investigated is part of a larger study investigating the roles of genetic and environmental factors in predicting response to stressful life events in a predominantly African-American, highly traumatized, urban population of low socioeconomic status and now counts over 4,000 participants3,37,38,39. All procedures in this study were approved by the Institutional Review Boards of the Emory University School of Medicine and Grady Memorial Hospital. The samples for the replication study of 56 female subjects were recruited as part of the larger Conte Center Study for the Psychobiology of Early-Life Trauma4,27. The study was approved by the Institutional Review Board of Emory University School of Medicine.
Psychometric instruments.
PTSD symptomatology was assessed by the mPSS as described previously3. The CAPS was used to assess current and lifetime PTSD diagnosis3. BDI was used to assess depressed mood in all individuals with the 21-item self report3. The CTQ was used as a continuous measure of childhood abuse and neglect in all individuals using the 28-item version of the CTQ40. For child abuse stratification, we used the subscales for sexual and physical abuse.
For the gene x environment interaction analyses, participants were dichotomized into two groups for each of the two categories of abuse (presence or absence of severe physical or sexual abuse according to CTQ). Cut-off scores of ≥13 for sexual abuse and ≥13 for physical abuse were used to define exposure to severe abuse in each of the categorized subjects. Finally, we created a 3-level categorical composite variable across the two abuse types, grouping participants into those with no severe exposure to any childhood abuse (scores less than cut-off for both types of abuse) and those with one or two exposures.
For the DNA methylation analyses, we divided participants into two groups for each of the three categories of abuse according to the presence or absence of moderate-to-severe physical, emotional or sexual abuse. Cut-off scores of ≥8 for sexual abuse, ≥13 for emotional abuse and ≥10 for physical abuse were used to define exposure to moderate to severe abuse in each of the categorized subjects. Finally, we created a categorical composite variable across the three abuse types, grouping participants into those with no moderate to severe exposure to any childhood abuse (scores less than cut-off for all types of abuse) and those with moderate to severe exposure to at least two types of abuse.
To allow for comparisons with previous studies3, we also used the TEI, which assesses lifetime history of trauma exposure to a range of traumatic events, including childhood sexual and physical abuse, and was assessed in all individuals. The TEI child abuse variable was highly concordant with the categorizations using the CTQ. To summarize the level of exposure to trauma other than child abuse, we summed the total number of different types of non-child abuse trauma exposure (TEI) reported by each subject. In this cohort, the different types of non–child abuse trauma were experience at an average of 22.6 ± 10.6 years (mean ± s.d.) of age, and we refer to this type of trauma as adult trauma.
DNA and RNA collection and extraction.
For genotyping, DNA was extracted either from saliva (Oragene DNA, DNA Genotek) or whole blood as described previously for the Grady trauma project3, and from whole blood for the Conte center sample4. For pyrosequencing, genomic DNA was extracted from peripheral blood using the Gentra Puregene Blood Kit (Qiagen) as described previously18. DNA quality and quantity was assessed by a NanoDrop 2000 Spectrophotometer (Thermo Scientific) and Quant-iT Picogreen (Invitrogen). For peripheral blood mRNA, blood was drawn between 8:00 and 9:00 a.m. into Tempus RNA tubes (Applied Biosystems) and stored at –20 °C until RNA extraction. RNA was isolated using the Versagene kit (Gentra Systems) and quantified using the Nanophotometer, and quality checks were performed on the Agilent Bioanalyzer as described previously15.
Genotyping.
In the Grady trauma cohort, genotype of rs1360780 was evaluated using TaqMan SNP genotyping assays (Applied Biosystems) and Sequenom i-plex assays (Sequenom). For imputation of SNPs in the FKBP5 locus, we used genotypes from the Illumina OmniExpress array. In the Conte center cohort and the human hippocampal progenitor cell line, rs1360780 was evaluated using a TaqMan SNP genotyping assay3.
Methylation analysis by bisulfite pyrosequencing.
Methylation analysis by pyrosequencing of bisulfite-treated genomic DNA was performed by varionostic GmbH (http://www.varionostic.de/). Genomic DNA was bisulfite converted using the EZ DNA Methylation Gold Kit (Zymo Research). Amplicons were generated from bisulfite DNA covering four intronic glucocorticoids-responsive elements, the CpG island around the TSS and one region upstream of the TSS (Fig. 4a and Supplementary Table 5). Sequencing was performed on the Q24 System with PyroMark Q24 analysis software in CpG (Qiagen). Data were analyzed using IBM SPSS Statistics, Version 17, as indicated. For subsequent analysis of DNA, we combined single CpG measures to bins, assuming that the DNA methylation around a transcription factor binding site is a functional unit. We divided the CpGs in the sequenced regions of FKBP5 into putative functional units according to their relative location to consensus GRE sites and chose the pyrosequencing primers accordingly. For example, in intron 7, three consensus GRE sites are described and their spatial relations to the CpG sites, the amplicon and sequenced regions are depicted in Supplementary Figure 2. CpGs in bin 1 are upstream of the 3 GREs, CpGs in bin 2 are surrounding and contained in the first consensus GRE, and the CpG in bin 3 is located in the third GRE. No CpGs were close to the second GRE.
Statistical methods.
The gene × environment interaction results with the mPSS score as outcome were obtained from 1,963 African-American subjects with complete FKBP5 genotype (rs1360780), CTQ and mPSS data and include individuals for which a gene × environment interaction had already been reported3, adding 1,194 new individuals. In this analysis, we restricted ethnicity to African American only, whereas, in the previous study, individuals from other ethnic backgrounds were also included (less than 10%). In addition, 519 individuals were examined using the CAPS for lifetime PTSD diagnosis, with 368 individuals not included in the previous study3. Child abuse was defined using the categorical CTQ child abuse variable as described above. The FKBP5 SNP rs1360780 was analyzed using a risk allele carrier model. A general linear model was used to analyze the effects on mPSS total score, with the 3-level categorical CTQ sexual and physical abuse variable described above, rs1360780, their interaction term, as well as age and sex as predictors. Controlling for current depressive symptom severity (using the BDI) and total types of trauma exposure (TEI) did not change the results. A logistic regression was used to analyze the effects of lifetime PTSD as assessed by the CAPS, with the categorical CTQ child abuse variable, rs1360780, in a risk allele carrier model, their interaction term, as well as age and sex as predictors. For post hoc analysis, a contingency table in the two risk genotype carrier groups tested for effects of severity of child abuse (CTQ child abuse 3 level variable) on the presence of lifetime PTSD.
SNPs within 304-kb segment containing the FKBP5 locus (Chr6, 35553493–35858276, version hg18) were selected from the OmniExpress Human SNP array (Illumina). Genotypes of these SNPs in 192 subjects from the Grady trauma projects were used to impute all variants in this locus detected in the 1000 Genomes projects19 version release June 2010 in Yorubans using Impute v.2 (ref. 41). The proximity of SNPs to GREs was assessed using data presented previously20, as well as ChIP-Seq data for the region of imputation from the ENCODE project (http://genome.ucsc.edu/cgi-bin/hgTrackUi?g=wgEncodeTfBindingSuper)42.
To examine DNA methylation of the FKBP5 locus, we selected an extreme subset of individuals from the Grady trauma project that had experienced at least both physical and sexual child abuse (N = 30) versus individuals that had not experienced any child abuse according to the TEI and CTQ and with no current or lifetime PTSD or depression (N = 45). For data analysis, the percentage of DNA methylation was summed for CGs lying in the same sequencing primer (Fig. 4a,b and Supplementary Table 5) and used as the dependent variable in a general linear model that included trauma exposure, FKBP5 risk allele carrier status, their interaction term, and age and sex as predictors. We corrected for multiple testing using a Bonferroni correction for the seven CG bins as well as three tests (main effect for trauma status, FKBP5 risk allele carrier status and their interaction) with an alpha level of 0.05/21 (P < 0.0023) considered as statistically significant. In a second type of analysis, DNA methylation was correlated to the severity of child abuse using the log10-transformed continuous CTQ score in both the Grady Trauma Project and the Conte center project.
Bonferroni corrections were applied three times. First, when testing the DNA methylation in the seven CpG bins as outcome for main SNP effects, main environment effect and interaction effects that is a total of 21 tests, second when testing correlations between DNA methylation of bin 2 in intron 7 with volumetric magnetic resonance imaging data. Here the correction was for the four brain regions (hippocampal head, body, tail and amygdala), but not each hemisphere, as the volumes across hemispheres are highly correlated. Third, we corrected for multiple testing when performing the pathway analyses for gene–expression where WikiPathways automatically corrects for the number of pathways tested. For these analyses, the corrected P values – Pcorr are presented.
RNA expression microarray and data analysis.
RNA was amplified and hybridized onto Illumina HT-12 v3.0 arrays (Illumina) as described previously15. Raw microarray scan files from the Illumina BeadScan confocal laser scanner were exported using the Illumina Beadstudio program and loaded into R. The data were transformed and normalized using the variance stabilizing normalization43. A total of 15,877 probes passing the filter criteria of Illumina probe detection P < 0.01 in 5% of the individuals were used for subsequent analysis. The expression profiles were further normalized using ComBat, an empirical Bayes method for batch correction. Association analysis was performed using general linear models in R. The significance of association was estimated by two-tailed P values using the ANOVA F test. Correlations were calculated using the Pearson correlations functions in R. Pathway analysis was done using WebGestalt, a WikiPathways tool with Bonferroni correction for multiple testing. Global mRNA expression data were assessed in individuals from the Grady trauma project. From a total of 396 with mRNA expression measures, we selected cases (N = 55, physical and sexual abuse, mPSS score > 20) and controls (N = 74, no childhood abuse, mPSS < 20). These groups were subsequently stratified by FKBP5 genotype into risk allele carrier status. To determine which transcripts are regulated by glucocorticoids, we regressed the gene expression profiles against the baseline cortisol levels, adjusting for age and gender in all 129 subjects. A total of 1,627 transcripts were significantly regulated by glucocorticoids with P < 0.01. For these transcripts, Pearson correlations between expression levels and baseline cortisol levels were calculated separately for cases and controls, further stratified by the FKBP5 genotype carrier model (that is risk versus protective allele). The correlations between the two genotype groups were converted into z score differences for cases and controls. z score differences of <1.96 indicate significance at P = 0.05.
Reporter plasmid construction and transient transfection.
The functional effect of differential methylation in intron 2 and intron 7 of FKBP5 was analyzed using a CpG-free luciferase reporter construct30 For intron 2, an 802-bp fragment containing the functional GRE was amplified from human genomic DNA with primer Intron2-F1 (containing a SpeI restriction site) and Intron2-R1 (containing a SbfI restriction site). The reporter plasmid was digested with SpeI and PstI to release the original CMV enhancer fragment. The PCR amplicon was digested with SbfI and SpeI, purified and subsequently ligated into the SpeI and PstI sites of the vector. The amplicon contains the single-nucleotide polymorphism rs1360780, and constructs with the A/T risk allele and the G/C protective allele were established (Supplementary Table 5). For intron 7, a 515-bp fragment was amplified from human genomic DNA with primer Intron7-F1 (containing a SpeI restriction site) and Intron7-R1 (containing a SbfI restriction site). The amplicon was ligated into the vector backbone as described above. All constructs were verified by sequencing (for additional information on primers and plasmids, see Supplementary Table 5). In vitro methylation of the reporter constructs by M.SssI CpG Methyltransferase (NEB) was carried out according to the manufacturer's instructions. Successful methylation was confirmed by restriction digest with PmlI (NEB), whereby linearization of the plasmids is blocked by CpG methylation. Methylated and unmethylated constructs were subsequently transfected into HEK-293 and HeLa cells (cultured in DMEM, supplemented with 10% fetal bovine serum, 1% sodium pyruvate and 1% antibiotic-antimycotic (vol/vol, Invitrogen)) using ExGen 500 in vitro Transfection Reagent (Fermentas) with Gaussia luciferase control vector for normalization of transfection efficacy. Firefly and Gaussia luciferase activity was measured in triplicates on a Tristar LB 941 luminometer with automatic injection system (Berthold Technologies). HEK 293 cells do not express the glucocorticoid receptor and are insensitive to dexamethasone stimulation. HeLa cells were transfected with the same constructs and successfully stimulated for 24 h with 50 nM dexamethasone. Data were analyzed using IBM SPSS Statistics, Version 17 as indicated.
TBP-DNA binding assay.
To validate the protein DNA interaction of TBP with the genomic sequence at rs1360780, we used the EpiQuik General Protein-DNA Binding Assay Kit (P-2004, Epigentek) with 60-mer double-stranded oligonucleotide probes (Supplementary Table 5). The single-stranded oligonucleotides were resuspended in an equimolar concentration in 1× TE buffer and annealed by heating the mixture to 95 °C for 10 min and gradually cooling to 20 °C. The assay was performed according to the protocol of the manufacturer with 5 ng of purified recombinant TBP protein (PR-703, Jena Biosciences). We used antibody to TBP (ab818, 1 μg ml−1) and horseradish peroxidase–conjugated antibody to mouse (ab6820, 0.5 μg ml−1) from Abcam. Absorbance was measured at 450 nm at 20 °C on a Tecan Genios Pro microplate reader (Tecan). The assays were performed in triplicates. Background absorbance was subtracted from the measurement reading. Specificity of the interaction was tested according to the manufacturer's protocol by addition of fivefold excess of non–biotin-labeled oligonucleotide to the reaction.
Chromatin conformation capture.
Chromatin conformation capture was carried out as described previously44. We used two human lymphoblastoid cell lines obtained from healthy individuals homozygous for the FKBP5 SNP rs1360780 GG and AA genotypes, respectively. Cells were cultured in RMPI with stable L-glutamine (Biochrom) supplemented with 10% fetal bovine serum and 1% antibiotic-antimycotic (Life Technologies). Crosslinking and cell lysis were performed as described44. Nuclei were digested using 400 U of EcoRI. Subsequent re-ligation, de-crosslinking and purification were conducted according to the protocol. DNA samples were adjusted to 20 ng μl−1 using the Quant-iT dsDNA BR Assay Kit and the Qubit 1.0 Fluorometer (Life Technologies). Digestion efficiency and sample purity was assessed as described previously44. Primers were designed with an anchor primer in the fragment containing the TSS and in potential interacting fragments in and around introns 2 and 7 of FKBP5 using Primer3. Quantitative PCR was carried out using the Roche LightCycler 480 SYBR Green I Master on a Roche LC480 according to the instructions of the manufacturer (Roche). A 176-kb BAC clone (RP11-282I23) containing the complete FKBP5 genomic sequence was obtained as a PCR control template from the BACPAC Resources Center of the Children's Hospital Oakland Research Institute. The BAC clone was cut with EcoRI and re-ligated by T4 DNA ligase. All primer pairs were tested on a standard curve of the BAC control library and yielded PCR efficiencies >1.7. The presence of a single PCR product was confirmed by agarose gel electrophoresis and melting curve analysis. Cycling conditions were: 95 °C for 5 min, 40 cycles of 95 °C for 15 s, 60 °C for 15 s, 72 °C for 20 s. Quantitative PCR data were normalized to GAPDH as a loading control and ERCC3 to control for interaction frequencies between samples. GAPDH cycling conditions were 95 °C for 5 min, 40 cycles of 95 °C for 10 s, 60 °C for 15 s, 72 °C for 20 s. ERCC3 cycling conditions were 95 °C for 5 min, 40 cycles of 95 °C for 15 s, 62 °C for 10 s, 72 °C for 25 s. Data analysis was carried out according to44 and is presented as relative crosslinking frequency. The primers used for the chromatin conformation capture interaction studies are listed in Supplementary Table 5.
Cell culture of multipotent human hippocampal progenitor cells.
Multipotent, human hippocampal progenitor cells (HPC03A/07, provided by ReNeuron) were grown in reduced modified media (RMM) as described previously29. To maintain proliferation, we added 10 ng ml−1 human bFGF (Peprotech), 20 ng ml−1 human EGF (Peprotech) and 100 nM 4-hydroxytamoxifen (4-OHT; Sigma-Aldrich). To induce differentiation, HPC03A/07 cells were washed and cultured in RMM media without EGF, bFGF and 4-OHT. To assess changes in FKBP5 methylation status in proliferating human hippocampal progenitor cells, we cultured HPC03A/07 cells in 25-cm2 tissue culture flasks (Nunclon) for 72 h with 1 μM dexamethasone. To assess changes in FKBP5 methylation under differentiation conditions, we treated HPC03A/07 cells for 72 h under proliferation conditions in RMM media containing EGF, bFGF and 4-OHT. After the 72-h proliferation phase, cells were washed twice for 15 min in RMM media without EGF, bFGF and 4-OHT, and subsequently cultured for another 7 d in media with 1 μM dexamethasone, but without growth factors. DNA was extracted using the Qiagen DNeasy Blood and Tissue kit (Qiagen) according to the manufacturer's instructions. Data were analyzed using IBM SPSS Statistics, Version 17, as indicated.
Ex vivo glucocorticoid receptor sensitivity.
To assess glucocorticoid receptor sensitivity, we used an ex vivo assay that estimates the degree of suppression of interleukin 6 secretion by the synthetic glucocorticoid, dexamethasone, in peripheral leukocytes, as described previously45,46.
Endocrine measures.
Serum cortisol levels were assessed in samples drawn at the same time as the whole blood samples used for DNA methylation analysis. Blood draws were performed between 8:00 and 9:00 a.m. in the Grady trauma project and at 12 p.m. in the replication sample. Cortisol levels were measured using a standard radioimmunoassay kit (Diagnostic System Laboratories).
Neuroimaging.
Individuals were scanned in the Biomedical Imaging Technology Center at Emory University School of Medicine using a 3.0T Siemens Magnetom TIM Trio Scanner (Siemens Medical Solutions). We acquired high-resolution T1-weighted image scans with isotropic 1-mm resolution using a three-dimensional spoiled gradient echo acquisition protocol with sagittal volume excitation to achieve contrast and resolution sufficient for structural analysis. All images were corrected for non-uniformity47, registered into standard stereotaxic space using the MNI template48, and normalized for signal intensity to harmonize gray-white matter contrast across subjects. For the amygdala and hippocampal segmentation, a segmentation protocol was employed with shown reliability and validity49.
Accession numbers.
Data from the microarray experiment are deposited at the GEO repository, GSE42002.
Accession codes
References
Caspi, A. & Moffitt, T.E. Gene-environment interactions in psychiatry: joining forces with neuroscience. Nat. Rev. Neurosci. 7, 583–590 (2006).
Caspi, A., Hariri, A.R., Holmes, A., Uher, R. & Moffitt, T.E. Genetic sensitivity to the environment: the case of the serotonin transporter gene and its implications for studying complex diseases and traits. Am. J. Psychiatry 167, 509–527 (2010).
Binder, E.B. et al. Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. J. Am. Med. Assoc. 299, 1291–1305 (2008).
Heim, C. et al. Effect of childhood trauma on adult depression and neuroendocrine function: sex-specific moderation by CRH receptor 1 gene. Front. Behav. Neurosci. 3, 41 (2009).
Xie, P. et al. Interaction of FKBP5 with childhood adversity on risk for post-traumatic stress disorder 3. Neuropsychopharmacology 35, 1684–1692 (2010).
Pratt, W.B. & Toft, D.O. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr. Rev. 18, 306–360 (1997).
Binder, E.B. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology 34 (suppl. 1), S186–S195 (2009).
Holsboer, F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology 23, 477–501 (2000).
Heim, C., Newport, D.J., Mletzko, T., Miller, A.H. & Nemeroff, C.B. The link between childhood trauma and depression: insights from HPA axis studies in humans. Psychoneuroendocrinology 33, 693–710 (2008).
Scammell, J.G., Denny, W.B., Valentine, D.L. & Smith, D.F. Overexpression of the FK506-binding immunophilin FKBP51 is the common cause of glucocorticoid resistance in three New World primates. Gen. Comp. Endocrinol. 124, 152–165 (2001).
Wochnik, G.M. et al. FK506-binding proteins 51 and 52 differentially regulate dynein interaction and nuclear translocation of the glucocorticoid receptor in mammalian cells. J. Biol. Chem. 280, 4609–4616 (2005).
Jääskeläinen, T., Makkonen, H. & Palvimo, J.J. Steroid up-regulation of FKBP51 and its role in hormone signaling 4. Curr. Opin. Pharmacol. 11, 326–331 (2011).
Appel, K. et al. Moderation of adult depression by a polymorphism in the FKBP5 gene and childhood physical abuse in the general population. Neuropsychopharmacology 36, 1982–1991 (2011).
Koenen, K.C. et al. Polymorphisms in FKBP5 are associated with peritraumatic dissociation in medically injured children. Mol. Psychiatry 10, 1058–1059 (2005).
Mehta, D. et al. Using polymorphisms in FKBP5 to define biologically distinct subtypes of post-traumatic stress disorder: evidence from endocrine and gene expression studies. Arch. Gen. Psychiatry 68, 901–910 (2011).
Roy, A., Gorodetsky, E., Yuan, Q., Goldman, D. & Enoch, M.A. Interaction of FKBP5, a stress-related gene, with childhood trauma increases the risk for attempting suicide. Neuropsychopharmacology 35, 1674–1683 (2010).
Zimmermann, P. et al. Interaction of FKBP5 gene variants and adverse life events in predicting depression onset: results from a 10-year prospective community study. Am. J. Psychiatry 168, 1107–1116 (2011).
Binder, E.B. et al. Polymorphisms in FKBP5 are associated with increased recurrence of depressive episodes and rapid response to antidepressant treatment 45. Nat. Genet. 36, 1319–1325 (2004).
1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010).
Paakinaho, V., Makkonen, H., Jaaskelainen, T. & Palvimo, J.J. Glucocorticoid receptor activates poised FKBP51 locus through long-distance interactions. Mol. Endocrinol. 24, 511–525 (2010).
Ising, M. et al. Polymorphisms in the FKBP5 gene region modulate recovery from psychosocial stress in healthy controls. Eur. J. Neurosci. 28, 389–398 (2008).
McGowan, P.O. et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat. Neurosci. 12, 342–348 (2009).
Murgatroyd, C. et al. Dynamic DNA methylation programs persistent adverse effects of early-life stress 8. Nat. Neurosci. 12, 1559–1566 (2009).
Weaver, I.C. et al. Epigenetic programming by maternal behavior. Nat. Neurosci. 7, 847–854 (2004).
Thomassin, H., Flavin, M., Espinas, M.L. & Grange, T. Glucocorticoid-induced DNA demethylation and gene memory during development. EMBO J. 20, 1974–1983 (2001).
Lee, R.S. et al. Chronic corticosterone exposure increases expression and decreases deoxyribonucleic acid methylation of Fkbp5 in mice. Endocrinology 151, 4332–4343 (2010).
Pace, T.W. et al. Increased peripheral NF-kappaB pathway activity in women with childhood abuse-related post-traumatic stress disorder. Brain Behav. Immun. 26, 13–17 (2011).
Sommershof, A. et al. Substantial reduction of naive and regulatory T cells following traumatic stress 1. Brain Behav. Immun. 23, 1117–1124 (2009).
Anacker, C. et al. Antidepressants increase human hippocampal neurogenesis by activating the glucocorticoid receptor. Mol. Psychiatry 16, 738–750 (2011).
Klug, M. & Rehli, M. Functional analysis of promoter CpG methylation using a CpG-free luciferase reporter vector. Epigenetics 1, 127–130 (2006).
Kelder, T. et al. Mining biological pathways using WikiPathways web services 3. PLoS ONE 4, e6447 (2009).
Danese, A. et al. Elevated inflammation levels in depressed adults with a history of childhood maltreatment 6. Arch. Gen. Psychiatry 65, 409–415 (2008).
Pace, T.W. et al. Increased stress-induced inflammatory responses in male patients with major depression and increased early life stress. Am. J. Psychiatry 163, 1630–1633 (2006).
Attwood, B.K. et al. Neuropsin cleaves EphB2 in the amygdala to control anxiety. Nature 473, 372–375 (2011).
Touma, C. et al. FK506 binding protein 5 shapes stress responsiveness: modulation of neuroendocrine reactivity and coping behavior 1. Biol. Psychiatry 70, 928–936 (2011).
O'Leary, J.C. III et al. A new anti-depressive strategy for the elderly: ablation of FKBP5/FKBP51. PLoS ONE 6, e24840 (2011).
Gillespie, C.F. et al. Trauma exposure and stress-related disorders in inner city primary care patients. Gen. Hosp. Psychiatry 31, 505–514 (2009).
Bradley, R.G. et al. Influence of child abuse on adult depression: moderation by the corticotropin-releasing hormone receptor gene. Arch. Gen. Psychiatry 65, 190–200 (2008).
Ressler, K.J. et al. Post-traumatic stress disorder is associated with PACAP and the PAC1 receptor. Nature 470, 492–497 (2011).
Fink, L.A., Bernstein, D., Handelsman, L., Foote, J. & Lovejoy, M. Initial reliability and validity of the childhood trauma interview: a new multidimensional measure of childhood interpersonal trauma. Am. J. Psychiatry 152, 1329–1335 (1995).
Howie, B.N., Donnelly, P. & Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies 1. PLoS Genet. 5, e1000529 (2009).
Birney, E. et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project 1. Nature 447, 799–816 (2007).
Huber, W., von, H.A., Sultmann, H., Poustka, A. & Vingron, M. Variance stabilization applied to microarray data calibration and to the quantification of differential expression 7. Bioinformatics 18 (suppl. 1), S96–S104 (2002).
Hagège, H. et al. Quantitative analysis of chromosome conformation capture assays (3C-qPCR). Nat. Protoc. 2, 1722–1733 (2007).
DeRijk, R.H., Petrides, J., Deuster, P., Gold, P.W. & Sternberg, E.M. Changes in corticosteroid sensitivity of peripheral blood lymphocytes after strenuous exercise in humans 2. J. Clin. Endocrinol. Metab. 81, 228–235 (1996).
Miller, G.E., Rohleder, N., Stetler, C. & Kirschbaum, C. Clinical depression and regulation of the inflammatory response during acute stress 5. Psychosom. Med. 67, 679–687 (2005).
Sled, J.G., Zijdenbos, A.P. & Evans, A.C. A nonparametric method for automatic correction of intensity nonuniformity in MRI data 4. IEEE Trans. Med. Imaging 17, 87–97 (1998).
Collins, D.L., Neelin, P., Peters, T.M. & Evans, A.C. Automatic 3D intersubject registration of MR volumetric data in standardized Talairach space. J. Comput. Assist. Tomogr. 18, 192–205 (1994).
Pruessner, J.C. et al. Volumetry of hippocampus and amygdala with high-resolution MRI and three-dimensional analysis software: minimizing the discrepancies between laboratories. Cereb. Cortex 10, 433–442 (2000).
Acknowledgements
We thank T. Mletzko, K. Hafner and N.C. Gassen for their help in acquiring and interpreting data, and P. Weber for data visualization. Special thanks to A.W. Graham, A. Brown, D. Crain and D. Cross for their assistance in recruiting subjects and managing the Grady Trauma Project. This work was supported by a European Research Council starting grant (grant #281338, GxE molmech), a grant from the National Alliance for Research in Schizophrenia and Affective Disorders and a grant from the Behrens Weise Stiftung to E.B.B., a grant from the National Institute of Mental Health (MH071538) to K.J.R., and a grant from the National Institute of Mental Health (MH58922) to C.B.N., C.M.P. and C.A. have received funding from the National Institute for Health Research Biomedical Research Centre for Mental Health, Institute of Psychiatry and South London and Maudsley National Health System Foundation Trust, a Clinician Scientist Fellowship from the UK Medical Research Council (G108/603) to C.M.P., and the Commission of European Communities 7th Framework Programme Collaborative Project Grant Agreement n 22963 (Mood Inflame), also to C.M.P., C.M.H. is supported in part by Public Health Service Grant UL1 RR025008 from the Clinical and Translational Science Award program, the US National Institutes of Health, the National Center for Research Resources and by a K Award (K01 MH073698–01, Neural Substrates of Depression Risk after Child Abuse).
Author information
Authors and Affiliations
Contributions
T.K. and E.B.B. designed the experiments, performed the luciferase assays and the genetic, methylation and expression analyses, analyzed the data, and wrote the initial version of the paper. D.M. performed the RNA expression experiments and data analyses and revised the paper. M.R.-H. performed the chromatin conformation capture experiments. C.A. and C.M.P. performed the cell-culture experiments with human hippocampal progenitor cells and revised the paper. T.W.W.P. performed ex vivo glucocorticoid receptor sensitivity experiments and revised the paper. J.C.P. analyzed magnetic resonance imaging data and revised the paper. E.B.B., F.H., K.J.R., K.B.M., H.S.M., B.B., C.B.N., C.M.H. and T.R. organized sample collection and collaborations, obtained funding, supervised data analyses, and revised the paper.
Corresponding authors
Ethics declarations
Competing interests
E.B.B., T.R. and F.H. hold a patent for the use of FKBP5 in antidepressant therapy (WO 2005/054500: FKBP5: a novel target for antidepressant therapy).
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–7 and Supplementary Tables 1–5 (PDF 293 kb)
Rights and permissions
About this article
Cite this article
Klengel, T., Mehta, D., Anacker, C. et al. Allele-specific FKBP5 DNA demethylation mediates gene–childhood trauma interactions. Nat Neurosci 16, 33–41 (2013). https://doi.org/10.1038/nn.3275
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nn.3275
This article is cited by
-
A DNA methylation signature in the stress driver gene Fkbp5 indicates a neuropathic component in chronic pain
Clinical Epigenetics (2023)
-
Association of APP gene polymorphisms and promoter methylation with essential hypertension in Guizhou: a case–control study
Human Genomics (2023)
-
Molecular pathways of major depressive disorder converge on the synapse
Molecular Psychiatry (2023)
-
Burning down the house: reinventing drug discovery in psychiatry for the development of targeted therapies
Molecular Psychiatry (2023)
-
Methylation and expression of glucocorticoid receptor exon-1 variants and FKBP5 in teenage suicide-completers
Translational Psychiatry (2023)
