

Guidelines & Standards

Evidence 2012;4(7): e1000025 doi: 10.4470/E1000025
Pubblicato: 27 novembre 2012
Copyright: © 2012 Moher et al. Questo è un articolo open-access, distribuito con licenza Creative Commons Attribution, che ne consente l’utilizzo, la distribuzione e la riproduzione su qualsiasi supporto esclusivamente per fini non commerciali, a condizione di riportare sempre autore e citazione originale.
Vedi anche: CONSORT Statement 2010

“Tutta la medicina dipende dalla trasparenza del reporting dei trial clinici” (1). Infatti, i trial controllati e randomizzati – randomized controlled trial (RCT) – ben disegnati e adeguatamente condotti forniscono le migliori prove di efficacia degli interventi sanitari, mentre quelli con metodologia inadeguata sono associati a bias e, in particolare, tendono a sovrastimare l’efficacia dei trattamenti (2-5). I risultati distorti di trial con disegno e reporting inadeguati possono determinare decisioni errate a tutti i livelli: dal trattamento del paziente individuale alle scelte nazionali di politica sanitaria.
Inoltre, la valutazione critica della qualità dei trial è possibile solo se il disegno, la conduzione e l’analisi dei RCT sono descritte in maniera completa e accurata. Lungi dall’essere trasparente, il reporting dei RCT è spesso incompleto (6-9), e comporta problemi conseguenti a una metodologia inadeguata (10-15).
Reporting incompleto e impreciso
Molte revisioni hanno documentato carenze nel reporting dei trial clinici: ad esempio, le informazioni sui metodi di assegnazione dei partecipanti a ciascun gruppo erano riportate solo nel 21% di 519 trial indicizzati in PubMed nel 2000 (16) e nel 34% dei 616 trial indicizzati nel 2006 (17). Analogamente, solo il 45% dei trial indicizzati in PubMed nel 200016 e il 53% nel 2006 (17) definivano un end-point primario, e solo il 27% nel 2000 e il 45% nel 2006 riportavano le metodologie utilizzate per stimare la dimensione del campione. Il reporting dei trial non solo è spesso incompleto, ma a volte è anche impreciso. Dei 119 trial in cui tutti i partecipanti erano analizzati secondo i gruppi di assegnazione originari (intention-to-treat analysis), 15 (13%) escludevano pazienti dall’analisi o non analizzavano tutti i pazienti nel gruppo di assegnazione (18). Numerose altre revisioni hanno riscontrato che un reporting inadeguato è più frequente nelle riviste specialistiche (16,19) e in quelle pubblicate in lingua non inglese (20,21).
Un’adeguata randomizzazione, riducendo il bias di selezione all’inizio del trial, è la componente fondamentale dei RCT di elevata qualità22. Una randomizzazione corretta richiede due step: la generazione di una sequenza di allocazione casuale e l’occultamento di tale sequenza ai ricercatori che arruolano i partecipanti (box 1) (2,23).
Box 1. Assegnazione dell’intervento: perché la randomizzazione è così importante? La metodologia utilizzata per assegnare i partecipanti a ciascun gruppo di intervento è un aspetto cruciale del disegno di un trial. L’assegnazione casuale è il metodo ideale, impiegato regolarmente con successo nei trial per oltre 50 anni (24). La randomizzazione ha tre vantaggi principali (25): in primo luogo, se applicata correttamente, elimina i bias di selezione, assicurando che tutti i fattori prognostici - sia noti che sconosciuti - si distribuiscano omogeneamente nel gruppo sperimentale e in quello di controllo. In assenza di randomizzazione, i confronti tra gli interventi possono risultare alterati, consapevolmente o meno, per la presenza del bias di selezione. In secondo luogo, l’assegnazione casuale consente di utilizzare la teoria della probabilità per esprimere la possibilità che ciascuna differenza di esito tra i gruppi di studio sia da attribuire all’efficacia del trattamento in studio (26). Infine, l’assegnazione casuale può in alcuni casi facilitare il blinding di ricercatori, partecipanti e valutatori degli esiti, grazie all’impiego di un placebo per ridurre i bias dopo l’assegnazione degli interventi (27). Di questi tre vantaggi, la prevenzione del bias di selezione all’inizio del trial è, indubbiamente, il più importante (27). Il successo della randomizzazione dipende da due aspetti strettamente correlati: un’adeguata generazione della sequenza di allocazione casuale e il suo occultamento sino all’assegnazione degli interventi (2,23). Un aspetto fondamentale è l’eventuale conoscenza o prevedibilità dell’assegnazione dei partecipanti ai gruppi di intervento da parte dei soggetti coinvolti nel trial (29). Il meccanismo di assegnazione degli interventi deve pertanto assicurare che chi arruola i partecipanti non conosca in anticipo a quale gruppo di intervento sarà assegnato il paziente successivo (occultamento della lista di randomizzazione) (2,23). Pertanto, se un’adeguata sequenza di allocazione consente di non prevedere le assegnazioni successive sulla base di quelle precedenti, un corretto occultamento della lista mantiene nascoste le assegnazioni successive. |
Purtroppo, nonostante il loro ruolo chiave, la descrizione delle metodologie utilizzate per l’assegnazione dei partecipanti agli interventi è generalmente inadeguata. Ad esempio, il 5% di 206 trial pubblicati in riviste di ostetricia e ginecologia erano in realtà studi non randomizzati (23). Questa stima è da considerarsi conservativa, poiché la maggior parte dei report attuali non descrive adeguatamente le metodologie di allocazione (20,23,30-33).
Come migliorare il reporting dei RCT: il CONSORT statement
DerSimonian et coll. hanno suggerito che “gli editori potrebbero migliorare sensibilmente il reporting di trial clinici, fornendo agli autori un elenco di item da riportare rigorosamente” (34). All’inizio degli anni ’90, due gruppi di editori, ricercatori e metodologi hanno pubblicato separatamente raccomandazioni per il reporting dei trial (35,36). A seguito di un invito di Rennie in un editoriale successivo, i due gruppi si sono incontrati sviluppando un set comune di indicazioni (37), dando vita al CONSORT (Consolidated Standards of Reporting Trials) Statement (38).
Il CONSORT Statement (o semplicemente CONSORT) comprende una checklist di item fondamentali che dovrebbero essere inclusi nel reporting dei RCT e un diagramma per documentare il flusso dei partecipanti nelle varie fasi di un trial. Pur essendo finalizzato a migliorare il reporting di trial a gruppi paralleli, numerosi item del CONSORT sono utili anche per altri disegni di trial: di non-inferiorità, di equivalenza, fattoriali, cluster e crossover. Per migliorare il reporting dei trial con questi disegni – così come il reporting di specifiche tipologie di dati (effetti avversi (42) ), di interventi (trattamenti non farmacologici (44), fitoterapia (44) ) e degli abstract (45), sono state pubblicate specifiche estensioni del CONSORT (39-41).
Il CONSORT ha l’obiettivo di fornire agli autori una guida per migliorare il reporting dei loro trial, rendendolo chiaro, completo e trasparente. Lettori, revisori ed editori possono utilizzare il CONSORT anche per la valutazione critica dei trial, ma è opportuno ricordare che il CONSORT non è stato elaborato con questa finalità. Molti item che, pur non definiti esplicitamente nel CONSORT, dovrebbero essere inclusi nel reporting di un trial: ad esempio le informazioni riguardanti l’approvazione del comitato etico, l’ottenimento del consenso informato da parte dei partecipanti, e, se rilevante, l’esistenza del comitato per la sicurezza dei dati e il monitoraggio. Inoltre, tutti gli altri aspetti di un trial che vengono menzionati devono essere adeguatamente riportati, come ad esempio i risultati di analisi di costo/efficacia (46-48).
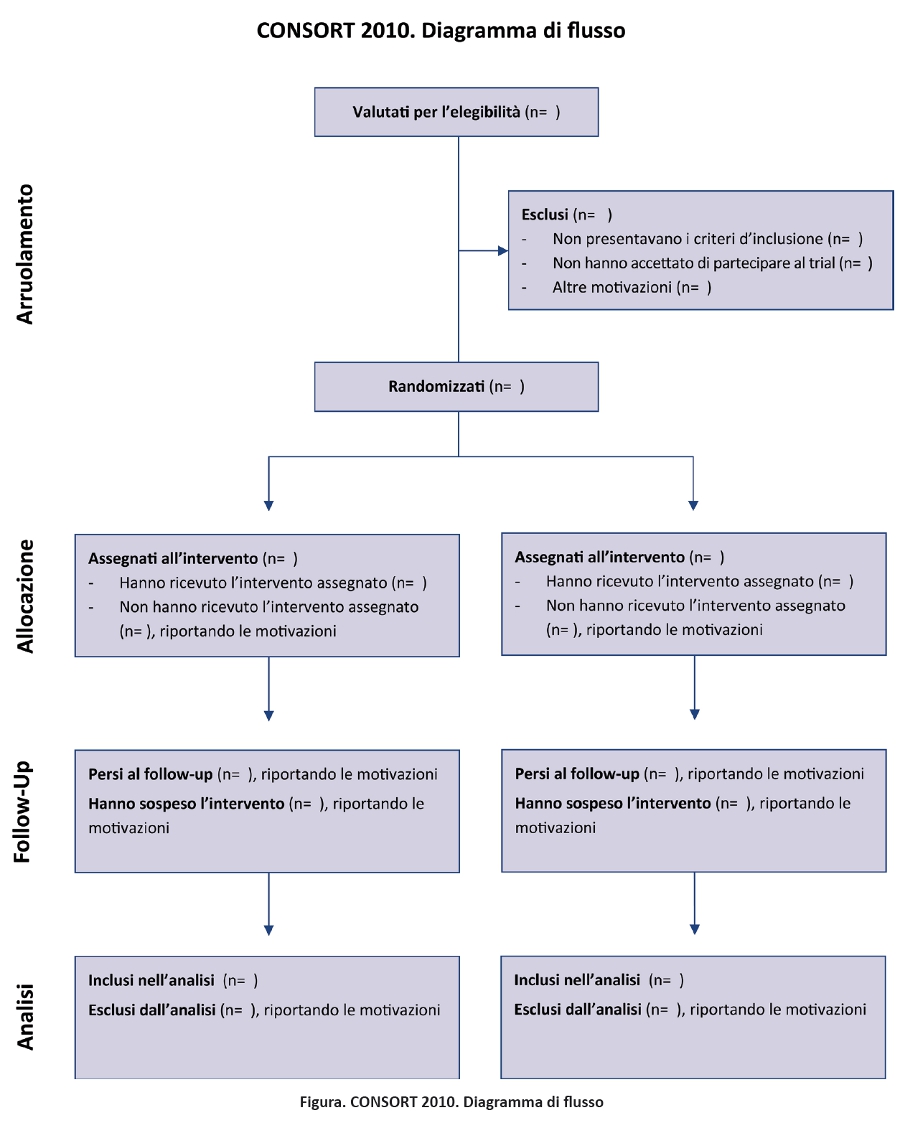
Dalla sua prima pubblicazione nel 1996, il CONSORT è stato adottato da oltre 400 riviste (www.consort-statement.org) e promosso da diversi gruppi editoriali, come l’International Committee of Medical Journal Editors (49), determinando un miglioramento della qualità dei reporting dei trial (17,50,51). Tuttavia, il CONSORT è un’iniziativa in corso e il CONSORT Statement viene revisionato periodicamente (3): l’ultima revisione risale al 2001 (52-54). Da allora le evidenze scientifiche per informare il CONSORT sono notevolmente aumentate e dati empirici hanno evidenziato l’importanza di nuove criticità metodologiche, come il reporting selettivo degli outcome (55-57). Il gruppo CONSORT si è riunito in Canada nel gennaio 2007 per revisionare il CONSORT statement 2001 e il relativo documento di spiegazione ed elaborazione. La checklist revisionata è riportata nella tabella 1 e il diagramma di flusso, non revisionato, nella figura 1 (52-54).
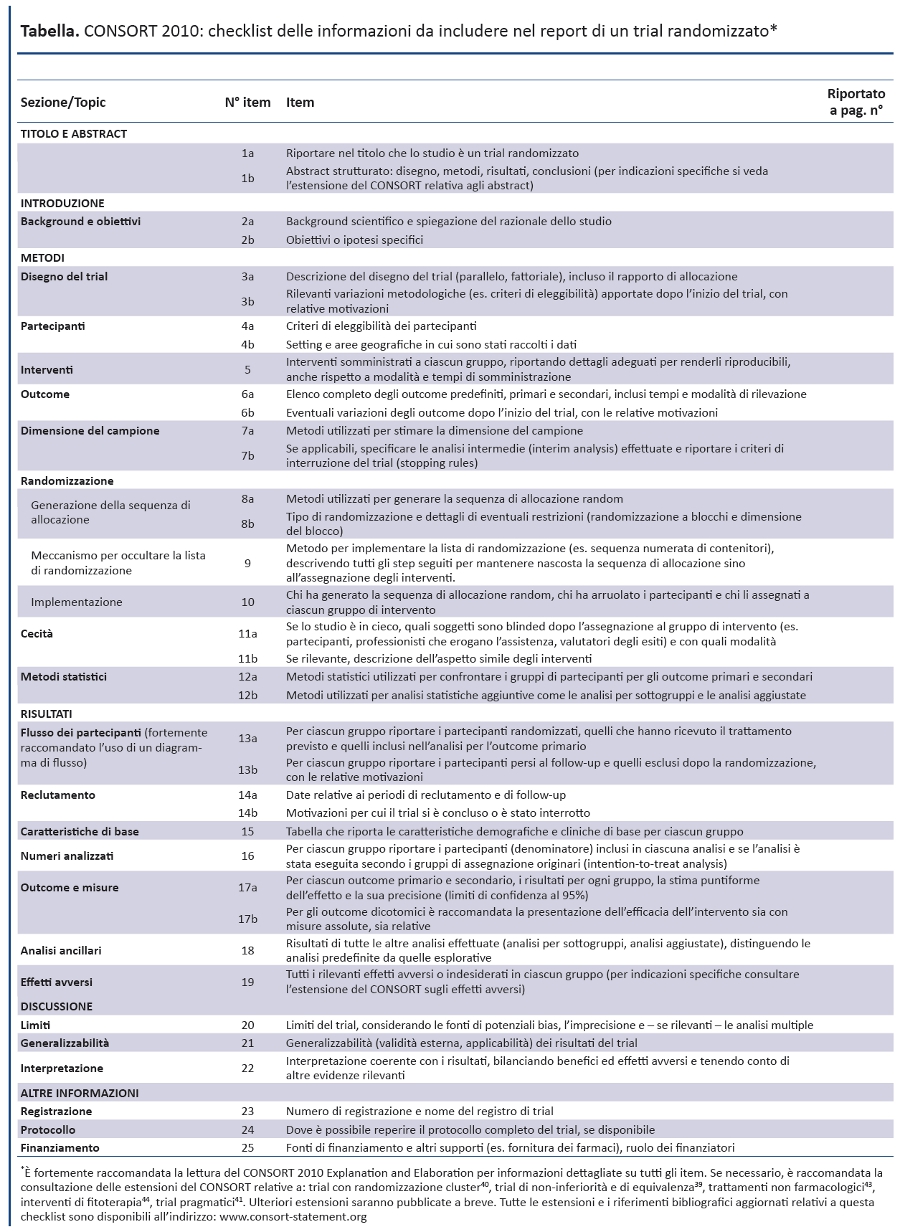
Il CONSORT Statement 2010: spiegazione ed elaborazione
Durante la revisione del CONSORT del 2001 è emerso che una spiegazione ed elaborazione dei singoli item avrebbero agevolato ricercatori e altri utenti nel reporting dei trial. Pertanto, insieme alla versione 2001 del CONSORT, è stato pubblicato un articolo di spiegazione ed elaborazione (58) che illustrava il background e il razionale scientifico di ogni item, riportando esempi pubblicati di reporting adeguato. Il razionale per la revisione dell’articolo di spiegazione ed elaborazione è simile a quello del CONSORT Statement. Descriviamo brevemente di seguito le principali considerazioni aggiunte e quelle eliminate della precedente versione dell’articolo di spiegazione ed elaborazione.
Le variazioni del CONSORT 2010 Spiegazione ed Elaborazione
Abbiamo apportato diverse modifiche sostanziali e alcune modifiche formali a questa versione del CONSORT 2010 Spiegazione ed Elaborazione (per i dettagli si veda la versione 2010 del CONSORT Statement (59) ). Alcune sono costituite da variazioni della checklist del CONSORT: ci sono tre item nuovi nella checklist del CONSORT 2010 – come l’item 24, che invita gli autori a indicare dove reperire il protocollo completo del trial. Abbiamo aggiornato alcune informazioni esistenti, riportando evidenze metodologiche più recenti e migliorato alcuni esempi. Abbiamo eliminato il glossario, ora disponibile sul sito web del CONSORT (www.consort-statement.org). Dove possibile, riportiamo ulteriori evidenze da studi empirici rilevanti. Molti libri eccellenti sui trial clinici offrono una discussione più ampia degli aspetti metodologici (60-62). Infine, per comodità, a volte utilizziamo la definizione “trattamenti” e “pazienti”, anche se non tutti gli interventi valutati nei RCT sono trattamenti e non tutti i partecipanti sono pazienti.
ITEM DELLA CHECKLIST
TITOLO E ABSTRACT
Item 1a. Riportare nel titolo che lo studio è un trial randomizzato.
Esempio. “Efficacia e sicurezza degli inalatori orali di nicotina per la disassuefazione al fumo: un trial clinico randomizzato in doppio cieco” (63).
Spiegazione. La capacità di identificare il report di un trial randomizzato in un database elettronico dipende in larga misura da come è stato indicizzato. È possibile che gli indicizzatori non classifichino un articolo come trial randomizzato se gli autori non riportano esplicitamente questa informazione (64). Per facilitare la corretta indicizzazione e l’identificazione di uno trial, gli autori dovrebbero utilizzare la parola “randomizzato” nel titolo, indicando che i partecipanti sono stati assegnati ai gruppi di confronto con modalità random.
Abstract strutturato: disegno, metodi, risultati, conclusioni (per indicazioni specifiche si veda l’estensione del CONSORT relativa agli abstract).
Per specifiche indicazioni si veda l’estensione del CONSORT relativa agli abstracts (45,65).
Spiegazione. E’ fondamentale che gli abstract siano chiari, trasparenti e sufficientemente dettagliati, perché spesso i lettori basano i loro giudizi esclusivamente su queste informazioni. Alcuni lettori utilizzano l’abstract come strumento di screening per decidere se leggere, o meno, l’articolo integrale. Inoltre, poiché non tutti i trial sono disponibili gratuitamente e non tutti i professionisti sanitari hanno accesso ai report completi dei trial, le decisioni cliniche vengono talora formulate sulla base delle informazioni contenute negli abstract (66).
Pertanto, l’abstract pubblicato su una rivista dovrebbe contenere informazioni adeguate e sufficienti da rappresentare una sintesi accurata delle metodologie e dei risultati del trial, secondo i vincoli editoriali e il formato della rivista.
Un abstract adeguatamente strutturato e ben redatto permette ai lettori di valutare rapidamente la rilevanza dei risultati e facilita il reperimento del trial nei database elettronici (67). L’abstract dovrebbe riflettere accuratamente i contenuti dell’articolo, senza includere informazioni che non compaiono nel testo integrale. Studi comparativi sull’accuratezza delle informazioni contenute negli abstract rispetto a quanto riportato nelle corrispondenti pubblicazioni integrali hanno rilevato dati incongruenti o mancanti rispetto all’articolo completo (68-71). Al contrario, omettere dall’abstract eventuali effetti avversi può indurre il lettore a interpretare erroneamente i risultati del trial (42,72).
Una recente estensione del CONSORT statement fornisce un elenco di item essenziali che gli autori devono includere pubblicando i risultati principali di un trial randomizzato in un abstract destinato ad una rivista o a un convegno (tabella 2) (45). Per il report di un trial randomizzato è fortemente raccomandato l’utilizzo di abstract strutturati, dove le informazioni sono organizzate in sezioni relative a disegno, conduzione, analisi e interpretazione (73). Alcuni studi hanno evidenziato che, rispetto agli abstract descrittivi, quelli strutturati hanno una qualità superiore (74,75) e permettono al lettore di identificare più facilmente le informazioni (76). In ogni caso, poiché numerose riviste hanno un proprio layout e impongono un limite di battute per gli abstract, non suggeriamo di modificare questi formati, ma ci limitiamo a raccomandare le informazioni da riportare.
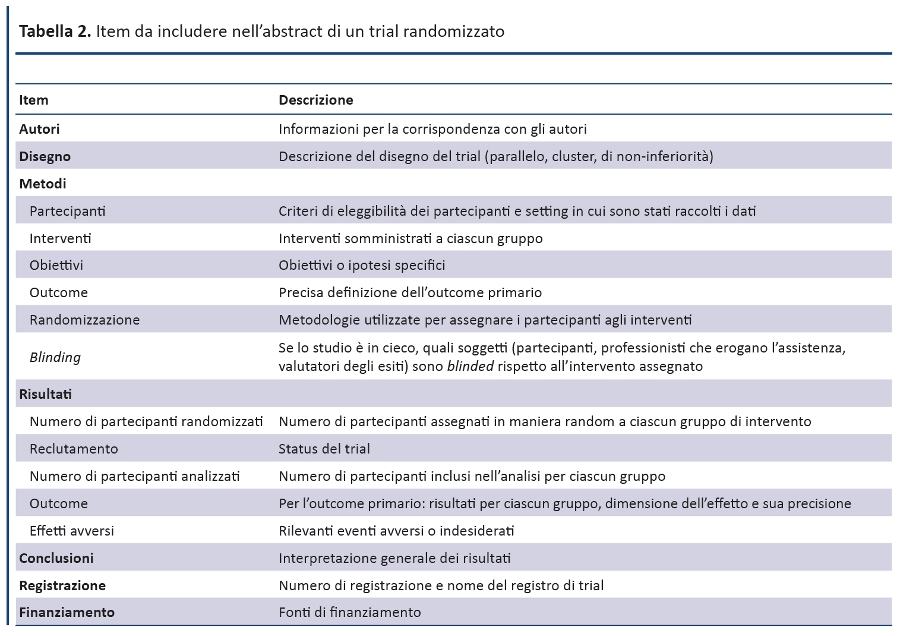
INTRODUZIONE
Item 2a. Background scientifico e spiegazione del razionale dello studio.
Esempio. “La chirurgia è il trattamento di prima scelta nei pazienti con carcinoma polmonare non a piccole cellule – non-small cell lung cancer (NSCLC) – di stadio I e II... Una meta-analisi sul NSCLC ha combinato i risultati di otto trial randomizzati che confrontavano l’intervento chirurgico rispetto alla combinazione chirurgia più chemioterapia adiuvante a base di cisplatino e ha mostrato un piccolo, ma non significativo (p = 0,08), beneficio assoluto di sopravvivenza di circa il 5% a 5 anni (dal 50% al 55%). Al momento della pianificazione del presente trial (metà degli anni ‘90), la chemioterapia adiuvante non era ancora il trattamento clinico standard... Il razionale scientifico per la chemioterapia neo-adiuvante è triplice: la regressione della neoplasia primaria potrebbe essere ottenuta in modo da semplificare o ridurre il successivo intervento chirurgico; micro-metastasi non identificate potrebbero essere considerate all’inizio del trattamento; si potrebbe inibire lo stimolo a neoplasie residue da parte di fattori di crescita rilasciati durante l’intervento chirurgico e la guarigione della ferita... il presente trial è stato quindi disegnato per confrontare, in pazienti con NSCLC resecabile, l’intervento chirurgico con tre cicli di chemioterapia a base di platino seguita dalla chirurgia in termini di sopravvivenza, qualità della vita, stadio di malattia, tasso di resecabilità, estensione della chirurgia, tempistiche e sedi delle recidive (77).”
Spiegazione. Di solito, l’introduzione è costituita da testo libero, dove gli autori spiegano il background e il razionale scientifico del trial e il suo schema generale. Può anche essere opportuno inserire nell’introduzione gli obiettivi del trial (item 2b). Il razionale può essere esplicativo (es., valutare la possibile influenza di un farmaco sulla funzione renale) o pragmatico (es., guidare la pratica clinica confrontando benefici ed effetti avversi di due interventi sanitari). Gli autori dovrebbero segnalare ogni evidenza di benefici ed effetti avversi degli interventi attivi inclusi in un trial e suggerire una spiegazione plausibile di come gli interventi dovrebbero funzionare, nel caso in cui non sia ovvio (78).
La Dichiarazione di Helsinki (79) sostiene che la ricerca biomedica che coinvolge soggetti umani dovrebbe essere basata su una conoscenza approfondita della letteratura scientifica, dal momento che non è etico esporre inutilmente gli esseri umani ai rischi della ricerca. Alcuni trial clinici sono risultati inutili perché il quesito valutato poteva essere risolto da una revisione sistematica della letteratura (80,81). Pertanto, la necessità di un nuovo trial deve essere giustificata nell’introduzione che, idealmente, dovrebbe includere un riferimento bibliografico a una revisione sistematica di trial simili già pubblicati, o una nota che specifichi che tali trial non sono ancora stati condotti (82).
Item 2b. Obiettivi o ipotesi specifici.
Esempio. “In questo studio abbiamo testato l’ipotesi che una gestione attiva del travaglio in donne nullipare dovrebbe: 1. Ridurre l’incidenza di taglio cesareo, 2. Ridurre l’incidenza di travaglio prolungato, 3. Non influenzare la soddisfazione materna durante l’esperienza del parto (83).”
Spiegazione. Gli obiettivi sono i quesiti a cui il trial intende dare una risposta, e spesso riguardano l’efficacia di uno specifico intervento terapeutico o preventivo. Le ipotesi sono i quesiti predefiniti che vengono valutati per poter raggiungere gli obiettivi. Le ipotesi sono più specifiche rispetto agli obiettivi e sottoposte a esplicita valutazione statistica; tuttavia, non sempre, obiettivi e ipotesi sono facilmente distinguibili. La maggior parte dei reporting di RCT fornisce adeguate informazioni relative a obiettivi e ipotesi del trial84.
METODI
Item 3a. Descrizione del disegno del trial (parallelo, fattoriale), incluso il rapporto di allocazione.
Esempio. “Studio multicentrico, stratificato (da 6 a 11 anni e da 12 a 17 anni di età, con randomizzazione non bilanciata [2:1]), in doppio cieco, controllato verso placebo, a gruppi paralleli condotto negli Stati Uniti (41 centri) (85).”
Spiegazione. Il termine “disegno” è spesso utilizzato per indicare tutti gli aspetti metodologici di pianificazione di un trial, ma può avere anche un’interpretazione più restrittiva. Molti aspetti specifici del disegno di un trial, come i dettagli di randomizzazione e cecità, sono trattati in altri item della checklist del CONSORT. In questa sezione sono presenti le informazioni sul tipo di trial, (es. a gruppi paralleli o fattoriale), il quadro concettuale (come la superiorità o la non-inferiorità), e altri aspetti correlati non analizzati in altri item della checklist.
Il CONSORT Statement si concentra principalmente sui trial con partecipanti randomizzati e assegnati individualmente a uno di due gruppi “paralleli”. In realtà, poco più del 50% dei trial pubblicati ha questo tipo di disegno (16). Tra i principali disegni alternativi ricordiamo i trial paralleli a bracci multipli, i trial con disegno crossover e fattoriali, i trial con randomizzazione cluster (40). Inoltre, se molti trial tendono, qualora esista, a dimostrare la superiorità di un nuovo intervento, altri ne valutano la non-inferiorità o l’equivalenza (39). È importante descrivere in modo chiaro questi aspetti dello studio, inclusa l’unità di randomizzazione (paziente, assistenza del medico di medicina generale, lesione), oltre a riportarli nell’abstract (item 1b).
Quando vengono utilizzati disegni di trial meno comuni, la scelta dovrebbe essere esplicitamente motivata, perché possono richiedere un campione di maggiori dimensioni, o analisi e interpretazioni più complesse.
Sebbene la maggior parte dei trial utilizzi la randomizzazione bilanciata (1:1 per due gruppi), si consiglia di esplicitare sempre il rapporto di allocazione. Per i trial farmacologici, può essere importante specificare anche la fase dello studio (I-IV).
Item 3b. Rilevanti variazioni metodologiche (es. criteri di eleggibilità) apportate dopo l’inizio del trial, con relative motivazioni.
Esempio. “I pazienti sono stati randomizzati e assegnati a uno dei sei gruppi paralleli, inizialmente secondo il rapporto 1:1:1:1:1:1, per ricevere uno dei cinque regimi di otamixaban [...] o un controllo attivo di eparina non frazionata [...] un comitato indipendente per il monitoraggio dei dati ha rivisto i dati relativi alla sicurezza del paziente non in cieco; non sono state eseguite analisi intermedie (interim analysis) per valutare l’efficacia. Durante il trial, il comitato ha raccomandato che il gruppo sottoposto alla dose più bassa di otamixaban (0,035 mg/kg/h) interrompesse il trattamento poiché la terapia anticoagulante risultava inadeguata. Sulla base di tale raccomandazione il protocollo è stato immediatamente modificato e i partecipanti sono stati successivamente randomizzati secondo il rapporto 2:2:2:2:1 rispettivamente ai 4 rimanenti gruppi di otamixaban e al controllo (86).”
Spiegazione. Se alcuni trial possono iniziare senza un protocollo stabilito (trial esplorativi), la maggior parte segue un protocollo che specifica nei dettagli le modalità di conduzione. Dal momento che è impossibile prevedere ogni eventuale cambiamento di situazioni, nel corso di un trial possono verificarsi deviazioni dal protocollo originale che richiederanno rilevanti variazioni metodologiche successive all’inizio del trial. Tali variazioni possono conseguire alla disponibilità di informazioni esterne derivanti da altri studi, a difficoltà finanziarie interne, oppure a un reclutamento inadeguato. Queste modifiche al protocollo dovrebbero essere apportate senza compromettere il blinding sugli outcome dei partecipanti. In alcuni trial, esiste un comitato indipendente di monitoraggio dei dati che può proporre variazioni del protocollo alla luce di dati non in cieco. Tali modifiche potrebbero influenzare i metodi di studio (es. variazioni di regimi di trattamento, criteri di eleggibilità, rapporto di randomizzazione o durata del follow-up) o la conduzione del trial (es. eliminazione di un centro con una scarsa qualità dei dati) (87).
Alcuni trial vengono progettati con un disegno “adattativo”. Non esiste una definizione universalmente accettata per questo disegno, ma in pratica lo si potrebbe definire “un disegno a più stadi che utilizza i dati raccolti per decidere come modificare gli aspetti dello studio, senza comprometterne validità e integrità” (88). Le modifiche, che solitamente riguardano la dimensione del campione e il numero dei bracci di trattamento, possono consentire decisioni più rapide e un più efficiente utilizzo delle risorse. Esistono, tuttavia, importanti aspetti etici, statistici e pratici da prendere in considerazione (89,90), e al fine di aiutare il lettore a interpretare i risultati, è essenziale riportare con estrema chiarezza se le modifiche apportate fanno parte del disegno dello studio o conseguono a mutate circostanze. Attualmente le variazioni metodologiche non vengono riportate in maniera adeguata: infatti, una revisione di confronti tra protocolli e rispettivi trial successivamente pubblicati ha evidenziato che circa la metà presenta inspiegabili discrepanze negli outcome primari (57), la randomizzazione, il blinding (91) e le analisi statistiche (92).
Item 4a. Criteri di eleggibilità dei partecipanti.
Esempio. “I partecipanti eleggibili erano gli adulti con infezione da HIV, di età ≥ 18 anni, che presentavano i criteri di eleggibilità per la terapia antiretrovirale secondo le linee guida nazionali del Malawi per il trattamento dell’HIV (stadio clinico III o IV OMS o qualsiasi stadio OMS con una conta di CD4 <250/mm3) e che all’inizio del trattamento avevano un BMI <18,5. I criteri di esclusione erano la gravidanza e l’allattamento, o partecipazione a un altro programma di nutrizione supplementare (93).”
Spiegazione. Per aiutare i lettori a interpretare lo studio, è necessario descrivere dettagliatamente i criteri di eleggibilità utilizzati per selezionare i partecipanti del trial. In particolare, una comprensione chiara di tali criteri è uno degli elementi indispensabili per stabilire a chi possono essere applicati i risultati di uno studio – cioè, la generalizzabilità del trial (applicabilità) e la rilevanza per la pratica clinica o la salute pubblica (item 21) (94). Altrettanto importante in questo senso è la descrizione del metodo di reclutamento, come l’invio selettivo o la partecipazione volontaria (es. attraverso la pubblicità). Dal momento che vengono applicati prima della randomizzazione, i criteri di eleggibilità non influenzano la validità interna di un trial, ma condizionano la sua validità esterna.
I criteri di eleggibilità tipici e ampiamente accettati dipendono dalla natura e dallo stadio della malattia oggetto di studio, mentre quelli di esclusione evitano l’arruolamento di partecipanti a rischio di eventi avversi dell’intervento in studio o per il rispetto di norme legali ed etiche. Il consenso informato dei partecipanti, ad esempio, viene tipicamente richiesto negli studi sperimentali. La distinzione tra criteri d’inclusione ed esclusione non è necessaria; lo stesso criterio può essere formulato per includere o escludere i partecipanti (95).
Nonostante la loro importanza, i criteri di eleggibilità spesso non vengono adeguatamente riportati. Ad esempio, otto trial pubblicati che hanno portato a segnalazioni cliniche da parte dei National Institutes of Health hanno descritto, in media, 31 criteri di eleggibilità nei loro protocolli, ma solo il 63% di tali criteri era riportato negli articoli pubblicati, e solo il 19% nelle segnalazioni cliniche (96). Carenze simili sono state riscontrate per i trial sull’HIV (97). Tra i 364 report di RCT in chirurgia, il 25% non ha indicato alcun criterio di eleggibilità (98).
Item 4b. Setting e aree geografiche in cui sono stati raccolti i dati.
Esempio. “Lo studio è stato eseguito presso il dipartimento di terapia antiretrovirale del Queen Elizabeth Central Hospital a Blantyre, Malawi, dal gennaio 2006 all’aprile 2007. Blantyre è la principale città commerciale del Malawi, con un 1.000.000 di abitanti e una prevalenza stimata di HIV del 27% negli adulti nel 2004 (93).”
Spiegazione. Oltre ai criteri di eleggibilità dei partecipanti (item 4a) e alla descrizione degli interventi (item 5), le informazioni sui setting e sulle aree geografiche sono fondamentali per valutare l’applicabilità di un trial. I partecipanti sono stati reclutati da setting di assistenza primaria, secondaria o terziaria o dalla comunità? Le istituzioni di assistenza sanitaria differiscono notevolmente in termini di organizzazione, esperienza e risorse e per quanto riguarda il rischio di base per la condizione in esame. Anche altri aspetti del setting (incluso il contesto sociale, economico e culturale e il clima) possono influenzare la validità esterna di uno studio.
Gli autori devono riportare il numero e la tipologia dei setting e descrivere i professionisti che erogano gli interventi sanitari. Devono inoltre riportare le aree geografiche in cui è stato condotto lo studio, inclusi il paese, se possibile la città, e l’ambiente circostante (es. la comunità, l’ambulatorio, l’ospedale o l’unità operativa). In particolare, dovrebbe essere chiaramente indicato se il trial è stato eseguito in uno o più centri (“trial multicentrici”). Questa descrizione dovrebbe fornire informazioni sufficienti per consentire ai lettori di valutare se i risultati dello studio possono essere rilevanti per il proprio setting assistenziale. Il contesto in cui viene condotto il trial può essere notevolmente diverso da quello in cui i suoi risultati saranno poi utilizzati per guidare la pratica clinica e le decisioni di politica sanitaria94,99. Gli autori devono fornire anche tutti gli altri dettagli relativi a setting e aree geografiche che potrebbero aver influenzato i risultati (es. problemi di viabilità e trasporti possono condizionare la partecipazione del paziente o determinare ritardi nella somministrazione degli interventi).
Item 5. Interventi somministrati a ciascun gruppo, riportando dettagli adeguati per renderli riproducibili, anche rispetto a modalità e tempi di somministrazione.
Esempi. “Nel trial POISE, i pazienti hanno ricevuto la prima dose del farmaco in studio (100 mg di metoprololo per via orale a rilascio prolungato) o del placebo 2-4 ore prima dell’intervento chirurgico. La somministrazione del farmaco richiedeva una frequenza cardiaca ≥ 50 bpm e una pressione sistolica ≥ 100 mmHg; questi parametri emodinamici sono stati monitorati prima di ogni somministrazione. Nelle prime 6 ore dopo l’intervento chirurgico, se in qualsiasi momento la frequenza cardiaca era ≥ 80 bpm e la pressione arteriosa sistolica ≥ 100 mmHg, i pazienti ricevevano la prima dose post-operatoria per via orale (100 mg di metoprololo a rilascio prolungato o placebo). Se il farmaco non era somministrato durante le prime 6 ore, i pazienti ricevevano la prima dose post-operatoria a 6 ore dall’intervento chirurgico. 12 ore dopo la prima dose post-operatoria, i pazienti iniziavano ad assumere 200 mg di metoprololo a rilascio prolungato per via orale o placebo, 1 volta/die per 30 giorni. Se la frequenza cardiaca del paziente era costantemente < 45 bpm o la pressione arteriosa sistolica scendeva sotto i 100 mmHg, il farmaco veniva sospeso sino ad un aumento della frequenza cardiaca o della pressione sistolica; il farmaco veniva, quindi, somministrato nuovamente al dosaggio di 100 mg/die. I pazienti in cui la frequenza cardiaca era costantemente 45-49 bpm e la pressione sistolica < 100 mmHg ritardavano l’assunzione del farmaco di 12 ore (100).”
“I pazienti erano randomizzati e assegnati a ricevere un tutore in neoprene su misura da indossare durante la notte oppure all’assistenza tradizionale. Il tutore, un’ortesi rigida raccomandata per uso esclusivamente notturno, ricopriva la base del pollice e il palmo, ma non il polso. I tutori erano stati realizzati da tre terapisti professionali esperti, che regolavano il tutore per ogni paziente in modo che il primo fotorecettore potesse essere aperto e il pollice posizionato dal lato opposto al primo dito lungo. I pazienti erano invitati a contattare il terapista se ritenevano necessaria una regolazione del tutore, in caso di aumento del dolore mentre indossavano il tutore, o di comparsa di effetti collaterali (es. erosione della pelle). Poiché in questa condizione non esiste nessun trattamento standard, i pazienti dei due gruppi (intervento e controllo) ricevevano cure tradizionali a discrezione del proprio medico di medicina generale o del reumatologo. Si è deciso di non utilizzare un placebo perché, secondo la nostra esperienza, nessun placebo per splintaggio è in grado di ottenere un blinding efficace dei pazienti (101).”
Spiegazione. Considerato che il medico che intende utilizzare l’intervento deve conoscerne esattamente le sue modalità di somministrazione nel trial, gli autori dovrebbero descrivere accuratamente ciascun intervento, inclusi quelli di controllo (102). Per un intervento farmacologico, le informazioni dovrebbero includere il nome del farmaco, la dose, le modalità di somministrazione (es. per via orale, endovenosa), le tempistiche e la durata della somministrazione, le condizioni in cui non è possibile erogare gli interventi e il regime di titolazione, se applicabile. Se il gruppo di controllo riceve “l’assistenza convenzionale” – usual care – è importante descrivere accuratamente da cosa è costituita. Se il gruppo sperimentale o quello di controllo riceve un intervento combinato, gli autori dovrebbero fornire sia una descrizione dettagliata di ciascun intervento, sia una spiegazione delle modalità con cui la combinazione degli interventi viene somministrata o interrotta, oltre ai fattori trigger per la loro introduzione, se applicabile.
Estensioni specifiche del CONSORT Statement riguardano il reporting di interventi non-farmacologici e fitoterapici ed evidenziano i dettagli specifici da prevedere (43,44), come il livello di competence professionale o i dettagli relativi alle modalità di standardizzazione degli interventi. Si raccomanda ai lettori di consultare le indicazioni per gli interventi non-farmacologici e fitoterapici, quando necessario.
Item 6a. Elenco completo degli outcome predefiniti, primari e secondari, inclusi tempi e modalità di rilevazione.
Esempio. “L’end-point primario relativo all’efficacia dell’intervento nella psoriasi era la percentuale di pazienti che raggiungeva un miglioramento del 75% dell’attività di malattia rispetto al valore di base dopo 12 settimane, secondo la misura del Psoriasis Area and Severity Index (PASI). Ulteriori analisi valutavano la variazione percentuale del punteggio PASI e il miglioramento delle lesioni psoriasiche (103).”
Spiegazione. Tutti i RCT valutano gli outcome (o end-point) per confrontare i risultati dei due gruppi di partecipanti. La maggior parte dei trial prevede diversi outcome, di cui alcuni hanno una maggiore rilevanza di altri. L’outcome primario è l’esito pre-definito considerato molto rilevante dai principali stakeholders (pazienti, policy-makers, medici, finanziatori) e viene utilizzato per stimare la dimensione del campione (item 7). La definizione di diversi outcome primari, anche se possibile, non è raccomandata perché può determinare problemi di interpretazione conseguenti alle analisi multiple (item 18 e 20). Gli outcome primari dovrebbero essere esplicitamente indicati come tali nel reporting di un RCT. Altri outcome di interesse sono quelli secondari (o accessori): gli outcome secondari possono essere diversi e spesso includono gli effetti avversi dell’intervento (item 19), che in realtà dovrebbero essere sempre considerati importanti, indipendentemente dal fatto che siano definiti primari o secondari.
Tutti gli outcome, primari e secondari, devono essere identificati e ben definiti nei dettagli per consentirne l’utilizzo ad altri ricercatori (102). Quando gli outcome vengono valutati in periodi diversi dopo la randomizzazione, gli autori dovrebbero indicare il time point pre-definito dell’outcome primario. Per molti interventi non farmacologici è utile specificare chi ha valutato gli esiti (es. se sono necessarie particolari competenze per farlo) e quanti erano i valutatori (43).
Qualora siano disponibili adeguate scale o linee guida, il loro utilizzo deve essere riportato (104,105), sia per migliorare la qualità delle misure, sia per consentire il confronto con studi simili (106). Ad esempio, la valutazione della qualità della vita può migliorare attraverso l’utilizzo di uno strumento validato (107). Gli autori dovrebbero sempre indicare la provenienza e le caratteristiche delle scale.
Oltre 70 outcome sono stati utilizzati in 196 RCT su farmaci anti-infiammatori non steroidei per l’artrite reumatoide (108), e 640 strumenti diversi sono stati utilizzati in 2.000 trial sulla schizofrenia, di cui 369 impiegati una sola volta (33). La valutazione di 149 di questi 2.000 trial ha dimostrato che l’utilizzo di scale non pubblicate in letteratura costituisce fonte di bias. Nei trial non farmacologici, un terzo delle definizioni di superiorità del trattamento, basato su scale non pubblicate, non sarebbe stato formulato se fosse stata utilizzata una scala pubblicata (109). Risultati analoghi sono stati riportati in altri campi (110,111). Solo il 45% di un gruppo di 519 RCT pubblicati nel 2000 specificava l’outcome primario16, analogamente al 53% di un gruppo simile di 614 RCT pubblicati nel 2006 (17).
Item 6b. Eventuali variazioni degli outcome dopo l’inizio del trial, con le relative motivazioni.
Esempio. “L’end-point primario originario era la mortalità per tutte le cause, ma, nel corso di un’analisi in cieco, il comitato di monitoraggio dei dati e della sicurezza ha riscontrato che la mortalità totale era inferiore a quella stimata e che lo studio non poteva essere completato con la dimensione del campione e la potenza definiti inizialmente. Il comitato direttivo ha pertanto deciso di utilizzare un duplice end-point primario: oltre alla mortalità per tutte le cause (end-point primario originario), la mortalità per tutte le cause insieme ai ricoveri per cause cardiovascolari (il primo end-point secondario predefinito) (112).”
Spiegazione. Esistono numerose motivazioni che richiedono di apportare variazioni al protocollo originale dello studio (item 24). Gli autori devono descrivere tutte le principali modifiche al protocollo, incluse quelle non previste ai criteri di inclusione, agli interventi, alle valutazioni, alla raccolta dei dati, ai metodi di analisi e agli outcome, informazioni che non sempre vengono riportate.
Come indicato in precedenza (item 6a), la maggior parte dei trial prende in considerazione diversi outcome, con il rischio di riportare i risultati solo per un sottogruppo selezionato (item 17); tale rischio dovrebbe essere prevenuto con la definizione preliminare e il reporting di outcome primari e secondari (item 6a). In alcuni trial, tuttavia, le circostanze richiedono una modifica delle modalità di valutazione di un outcome o addirittura, come nel precedente esempio, la modifica dell’outcome primario. Ad esempio, evidenze derivanti da altri trial o revisioni sistematiche possono suggerire che l’end-point potrebbe non essere appropriato; oppure il reclutamento dei partecipanti o l’incidenza dell’evento nel trial potrebbero essere inferiori al previsto (112). La variazione di un end-point sulla base di dati non in cieco è molto più problematica, anche se può essere specificata nel contesto di un trial con disegno adattativo88. Gli autori devono identificare e motivare tali modifiche, oltre che così riportare e spiegare eventuali variazioni agli outcome apportate dopo l’inizio del trial.
Un confronto tra protocolli e pubblicazioni di 102 trial randomizzati ha riscontrato che il 62% dei trial aveva almeno un outcome primario modificato, introdotto, oppure omesso rispetto al protocollo originale (55). Differenze negli outcome primari erano anche evidenti tra protocolli e pubblicazioni nel 40% di 48 trial finanziati dal Canadian Institutes of Health Research (113). Nessuno dei 150 report di trial successivamente pubblicati riportava, e tanto meno spiegava, le modifiche apportate al protocollo. Analoghi risultati provenienti da altri studi sono stati recentemente menzionati da una revisione sistematica di studi empirici che analizza i bias nel reporting degli outcome (57).
Item 7a. Metodi utilizzati per stimare la dimensione del campione.
Esempi. “Per rilevare una riduzione della degenza post-operatoria di 3 giorni (deviazione standard 5 giorni), in accordo con lo studio di Lobo et al. (17) con un livello di significatività del 5% e una potenza dell’80%, è stato stimato un campione di 50 pazienti per gruppo, considerando un’incidenza di abbandono del 10%. Per reclutare questo numero di pazienti è stato previsto un periodo di 12 mesi (114).”
“Sulla base di un’incidenza attesa dell’end-point primario composito del 11% dopo 2,25 anni nel gruppo placebo, abbiamo stimato necessari 950 eventi dell’end-point primario e un campione di 9.650 pazienti, con una potenza di 90% per rilevare una differenza significativa tra ivabradina e placebo, corrispondente a una riduzione relativa del 19% del rischio (errore a pari al 5%). Inizialmente abbiamo disegnato un trial event-driven, stabilendo di interromperlo una volta rilevati 950 end-point primari. Tuttavia, l’incidenza dell’end-point primario è stata superiore al previsto, forse per le caratteristiche di base dei pazienti reclutati, che presentavano un rischio più elevato rispetto a quello atteso (es. minore percentuale di pazienti classe NYHA I e incidenza più elevata di diabete e ipertensione). Abbiamo calcolato che nel momento in cui si fossero verificati 950 end-point primari, i pazienti inclusi più recentemente sarebbero stati trattati solo per circa 3 mesi. Di conseguenza, nel gennaio 2007, il comitato esecutivo ha deciso di modificare lo studio da event-driven a time-driven e di proseguire sino a 12 mesi il follow-up degli ultimi pazienti randomizzati. Questa variazione non ha modificato la durata di 3 anni prevista dello studio (115).”
Spiegazione. Per ragioni scientifiche ed etiche, la dimensione del campione di un trial deve essere accuratamente stimata, bilanciando valutazioni cliniche e statistiche. Idealmente, uno studio deve essere sufficientemente numeroso da avere un’elevata probabilità (potenza) di rilevare come statisticamente significativa una differenza clinicamente importante, se questa esiste realmente. La dimensione dell’effetto considerato importante è inversamente proporzionale alla dimensione del campione necessario per rilevarla: in altre parole per rilevare piccole differenze sono necessari campioni molto numerosi. Gli elementi per calcolare la dimensione del campione sono (1) l’incidenza degli outcome stimati in ciascun gruppo (che implica la differenza target clinicamente rilevante tra i gruppi di intervento); (2) l’errore a (tipo I); (3) la potenza statistica, oppure l’errore ß (tipo II); (4) per gli outcome continui, la deviazione standard (116). L’interazione di questi elementi e il loro reporting saranno diversi per i trial co